
Abstract
Rifampin has been a cornerstone of tuberculosis (TB) treatment since its introduction. The rise of multidrug-resistant and extensively drug-resistant TB makes the development of novel therapeutics effective against these strains an urgent need. Site-specific mutations in the target enzyme of rifampin, RNA polymerase (RNAP) comprises the majority (~97%) of rifamycin-resistant (RifR) strains of Mycobacterium tuberculosis (MTB). To identify novel inhibitors of bacterial RNAP, an in vitro plasmid-based transcription assay that uses malachite green (MG) to detect transcribed RNA containing MG aptamers was developed. This assay was optimized in a 384-well plate format and used to screen 150,000 compounds against an Escherichia coli homolog of the most clinically relevant RifR RNAP (βS531L) containing a mutation (β′V408G) that compensates for the fitness defect of this RifR mutant. Following confirmation and concentration-response studies, 10 compounds were identified with similar in vitro inhibition values across a panel of wild-type and RifR E. coli and MTB RNAPs. Four compounds identified from the screen are active against MTB in culture at concentrations below 200 µM. Initial follow-up has resulted in the elimination of one scaffold due to potential pan-assay interference.
Introduction
Tuberculosis (TB), caused by the pathogen Mycobacterium tuberculosis, is one of the largest global health problems. TB exists in two forms, latent and active, with an estimated one-third of the global population infected with the latent form. There are an estimated 8 to 10 million new cases of TB each year (10.4 million new cases in 2015). TB is one of the leading causes of death worldwide, killing 1.8 million people (1.4 million HIV-negative and 0.4 million HIV-positive) globally in 2015, rivaling HIV-associated deaths (1.1 million) as the leading cause of death from infectious diseases. 1 Global treatment success rates for newly diagnosed TB were 83% in 2014. 2 The current treatment regimen for TB is a 6- to 9-month time course of four different first-line anti-TB drugs: rifampin (RMP), isoniazid, ethambutol, and pyrazinamide. However, there are increasing numbers of cases of drug-resistant TB, with an estimated 480,000 new cases of multidrug-resistant TB (MDR-TB; resistant to RMP and isoniazid) in 2015, of which 10% is estimated to be extensively drug-resistant TB (XDR-TB), having additional resistance to some second-line anti-TB drugs. Globally, only 50% of MDR-TB patients were successfully treated. It is clear that novel drugs are still needed for successful treatment of individuals with MDR- or XDR-TB.
Despite this clear and pressing need, only two TB drugs with novel mechanisms have been introduced in the past 50 years (bedaquiline and delamanid), and MTB strains resistant to both drugs have already appeared in the clinic.2,3 Reinvestigating previously effective drug targets has, in some cases, proven to be successful in antibiotic drug discovery.4,5 The lack of new drugs effective against MDR- and XDR-TB makes developing novel inhibitors of previously successful targets an attractive approach.
RMP, one of the first-line anti-TB drugs, was first introduced in the 1960s and is bactericidal by inhibiting the bacterial DNA-dependent RNA polymerase (RNAP). 6 However, rifamycin-resistant (RifR) mutations in the RNAP arise at a high rate (~10−8), necessitating the use of RMP in combination with other drugs to minimize resistance development. 7 Mutations within an 81-bp core region of the rpoB gene, known as the RMP resistance determining region (RRDR), account for ~96% of all RifR strains of MTB. 7 Mutations at three residues in the RRDR (S450, H445, D435) account for ~84% of RifR strains, and the most clinically prevalent mutation (S450L) accounts for ~45% of RifR MTB. These mutations dramatically reduce the affinity of RNAP for rifamycins. 8 This suggests that novel inhibitors of RNAP that bind in sites other than the RRDR should be effective against RifR TB. To identify novel inhibitors of RifR RNAP, we sought to perform a high-throughput screen (HTS) of ~150,000 small molecules against the E. coli homolog of the most clinically prevalent RifR MTB RNAP, βS450L (E. coli βS531L). Recent studies have shown the S450L RifR mutant to have a fitness defect that in many clinical strains is compensated for by specific secondary mutations in rpoC.9,10 The most effective compensating mutation is V483G of the β′ subunit. To reflect what is likely the most prevalent RifR RNAP in RifR, MDR- and XDR-MTB, the E. coli RNAP βS531L/β′V408G (corresponding to βS450L/β′V483G in the MTB numbering) was used for the HTS.
Previous screens for inhibitors of RNAP have used radiolabeled or fluorescent nucleotide analogs.11,12 Another approach involved detection of the RNAP core enzyme–σ factor interaction (holoenzyme assembly).13,14 HTS using nucleotide analogs can be cost prohibitive, or impractical in the case of radiolabeled NTPs, whereas detecting the RNAP–σ interaction could significantly limit the potential modes of RNAP inhibition. Therefore, we developed a robust enzymatic assay in which an inexpensive, commercially available dye (MG) could be used for detection of RNA transcribed by RNAP from a plasmid template ( Fig. 1A ). 15 The product RNA contains repeats of a 38-nucleotide RNA aptamer that binds MG and results in an ~2000-fold increase in MG fluorescence. This assay was used to screen E. coli RNAP βS531L/β′V408G to discover compounds for potential drug development against MDR- and XDR-TB.
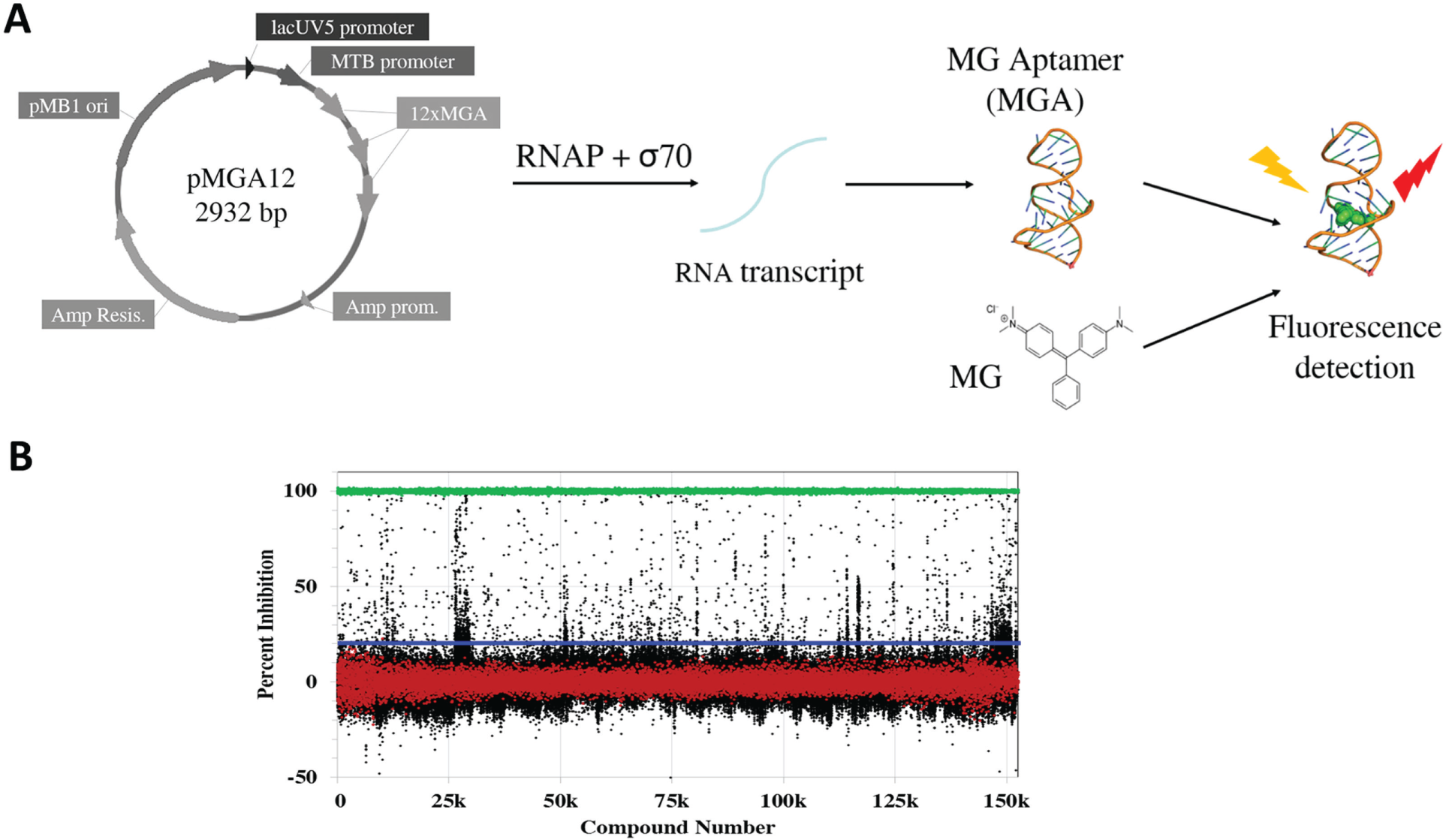
(
Materials and Methods
Reagents
All reagents were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise specified. Nucleoside triphosphates were purchased from Chem-Impex International (Wood Dale, IL). QIAprep Spin Miniprep Kit, Giga Kit, QIAquick Gel Extraction Kit, QIAquick Nucleotide Removal Kit, and Ni-NTA agarose were from Qiagen (Valencia, CA). Yeast extract, bactotryptone, kanamycin, DTT, Corning microplates, and Amicon Centrifugal Filter Units were purchased from Fisher Scientific (Hampton, NH). Carbenicillin and isopropyl β-D-1-thiogalactopyranoside (IPTG) were purchased from Gold Biotechnology (St. Louis, MO). Chloramphenicol was purchased from Tokyo Chemical Industry (Tokyo, Japan). Taq DNA polymerase, T4 DNA ligase, Antarctic phosphatase, deoxynucleoside triphosphates (dNTPs), and all restriction enzymes were purchased from New England Biolabs (Ipswich, MA). PfuUltra high-fidelity DNA polymerase was purchased from Agilent Technologies (Santa Clara, CA). pCR2.1-TOPO Cloning Kit, Quant-iT RiboGreen RNA reagent, and all synthetic oligonucleotides were purchased from Invitrogen (Carlsbad, CA). Oligonucleotide primers used in this study are listed in
Strains and Plasmids
MTB H37Rv and Vero cells (ATCC) were used for minimum inhibitory concentration (MIC) and cytotoxicity assays, respectively. Plasmid construct pVS10 for wild-type (WT) E. coli RNAP expression was a generous gift from Irina Artsimovitch (Ohio State University). The preparation of pVS10 E. coli RNAP expression plasmids for β subunit mutations D516V, H526Y, S531L was previously described. 16 The pU6+19 vector containing the four MG aptamer (MGA) repeats (pU6+19-4xMGA) was a generous gift from Dr. Marit Nilsen-Hamilton (Iowa State University). 17 The pFPV27-prrn plasmid was a generous gift from Dr. Jaya Tyagi (India Institute of Medical Sciences). 18 Other plasmids used in this study were prepared as described below.
Small-Molecule Library
A 150,554 compound library was screened at the Center for Chemical Genomics (CCG; University of Michigan, Ann Arbor). The library consisted of compounds from the Chemical Diversity set (120,000 compounds), Maybridge HitFinder, MicroSource Spectrum, and NCC BioFocus libraries.
Construction of Reporter Plasmid
The construction and optimization of the reporter plasmid is described in the supplemental material.
RNAP Expression Vectors
An expression plasmid for E. coli S531L RNAP with the compensatory mutation β′V408G was prepared from the expression plasmid pVS10(S531L) as follows. The wild-type E. coli rpoC gene was amplified from pVS10 using oligonucleotides ECrpoC-Fwd and ECrpoC-Rev and subcloned using the TOPO Cloning Kit (pCR2.1-TOPO). The compensatory mutation V408G (GTT–GGT) was introduced to rpoC via two-stage site-directed mutagenesis with complementary oligonucleotides ECrpoCMut-Fwd and ECrpoCMut-Rev. The mutant rpoC gene and pVS10 (S531L) were restricted with SbfI and SacII and ligated with T4 DNA ligase to prepare pVS10 (S531L/V408G). Expression vector pET15b-Sig70 used for expression of E. coli σ70 was previously prepared (V. Molodtsov and K. S. Murakami, unpublished data).
MTB RNAP expression vectors pACYCDuet-rpoA-rpoZ and pETDuet-rpoB-rpoC were prepared using the same approach as used by Banerjee et al.,
19
amplifying and inserting the MTB genes using appropriate primers and restriction enzymes (
Expression and Purification of E. coli and MTB RNAPs
Expressions and purifications of E. coli and MTB RNAPs were conducted as previously described, 21 with a few modifications as described in the supplemental material.
Expression and Purification of N-His6–E. coli σ70
E. coli BL21(DE3) containing pET15b-Sig70 (WT rpoD) were grown in 3 L 2xTY at 30 °C until OD600 ~ 0.5, then induced with 1 mM IPTG and allowed to grow for an additional 2 h at 22 °C. Cells were harvested by centrifugation and resuspended in 30 mL σ70 lysis buffer (20 mM Tris-HCl, 0.4 M NaCl, 1 mM EDTA, 2 mM phenylmethane sulfonyl fluoride [PMSF], 5 mM β-ME, 5% glycerol, pH = 8.0 at 4 °C) and then sonicated on ice. The lysate was cleared by centrifugation (35,000g, 30 min, 4 °C), and the supernatant (30 mL) was applied to a 10-mL Ni2+ column equilibrated with σ70 lysis buffer + 20 mM imidazole and then washed with 20 column volumes (CVs) of the same buffer. Protein fractions (25 mL) were eluted with 50-mL σ70 elution buffer (10 mM Tris-HCl, pH 8.0 at 4 °C, 200 mM NaCl, 5% glycerol, 0.1 mM EDTA, 5 mM β-ME, 200 mM imidazole). Next, the protein was diluted to reduce the concentration of NaCl to 100 mM and applied to a 5-mL Q Sepharose column equilibrated with σ70 elution buffer (without imidazole) and a linear gradient of 0.1 to 0.6 M NaCl over 20 CVs. Fractions containing σ70 were determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, combined together (60 mg in 20 mL), and flash frozen in liquid N2 until further use.
Expression and Purification of MTB σA
E. coli BL21(DE3) containing pMCSG7-SigA were grown in 2 L 2xTY at 37 °C until OD600 = 0.4 to 0.6, then expression was induced with 1 mM IPTG and grown at 16 °C for 16 to 20 h. Cells were pelleted and resuspended in σ70 lysis buffer and supplemented with 2 mM PMSF. Cells were sonicated, and lysate was cleared by centrifugation (21,000g, 40 min, 4 °C). Supernatant was incubated with 2-mL Ni-NTA agarose and incubated at 4 °C with shaking for 1 h. The mixture was then applied to a column and washed as follows: 5 CVs, σ70 lysis buffer; 8 CVs, σ70 lysis buffer + 15 mM imidazole; 10 CVs, σ70 lysis buffer + 100 mM imidazole. Eluted protein was concentrated using a 10 kDa MWCO Amicon Ultra-15 Centrifugal Filter Unit, filtered, and applied to a HiPrep 16/60 Sephacryl S-200 HR equilibrated with TGED containing 200 mM NaCl. Protein was pooled and concentrated and then flash frozen and stored at −80 °C. Protein can be stored as is or containing glycerol (final concentration of 25% glycerol was used).
In Vitro Transcription Assay
The activity of E. coli RNAP was measured via plasmid-based, in vitro transcription using pMGA12. A 50 µL reaction was prepared in Corning 96-well half-area black flat-bottom polystyrene NBS microplates. In each well, 24 µL of a 2x E. coli reaction buffer solution (80 mM Tris-HCl [pH = 7.5 at 23 °C], 100 mM KCl, 20 mM MgCl2, 0.02% Triton X-100, 16 mM DTT, 20 nM pMGA12, 1 mM NTPs) was added, 2 µL of DMSO or compound in DMSO, followed by 24 µL of a 2x RNAP solution (80 nM RNAP, 240 nM σ70). Reactions were incubated at 37 °C for 2 h then halted by incubating on ice and adding 50 µL of cold 60 µM MG in water. After 5 min on ice, MG fluorescence was detected at excitation and emission wavelengths of 628 nm and 660 nm, respectively. MTB RNAP transcription assays were done the same as E. coli RNAP; however, the following 2x MTB reaction buffer solution was used: 40 mM Tris-HCl (pH = 8.0 at 37 °C), 20 mM MgCl2, 300 mM potassium L-glutamate, 15% glycerol.
High-Throughput Bacterial Transcription Assay
The transcription assay described above was modified to 384-well plate format, with a reaction volume of 20 µL. A 2x E. coli reaction buffer was added (10 µL) to each well of 384-shallow well microplates (ProxiPlate-384 Plus F) using a multidrop dispenser (Thermo Scientific, Waltham, MA). Compounds or DMSO were then added (20 µµM final, n = 1, 0.2 µL) to the appropriate wells using a Biomek FX HDR pintool instrument (Beckman, Fullerton, CA). TGED containing 50 mM NaCl and 0.01% Triton X-100 was added (10 µL) to the appropriate wells for a positive control via multidrop dispenser. A 2x RNAP solution (80 nM βS531L/β′V408G E. coli RNAP core, 240 nM E. coli σ70 in TGED containing 50 mM NaCl and 0.01% Triton X-100 was added to the appropriate wells (10 µL) via multidrop dispenser for a reaction volume of 20 µL. The plates were incubated at 37 °C for 2 h and then placed on ice for 10 min. A solution of 150 µM MG was added (5 µL) to each well. The plates were incubated on ice for 10 min before measuring fluorescence in an EnVision Multilabel plate reader (PerkinElmer) using a Cy5 620/10 filter for excitation and a LANCE 665/7.5 filter for emission.
Confirmation and Counterscreens
Compounds identified from the primary HTS were tested against a series of control screens. First, compounds were retested (n = 3) in the assay following the protocol above, except compounds were added to empty plates using a mosquito X1 (TTP Labtech) prior to addition of buffer and RNAP solutions. These compounds were also tested for interference of MG•MGA fluorescence. This was done by adding 20-μL of preformed RNA (1x E. coli reaction buffer solution and 1x E. coli RNAP solution incubated at 37 °C for 2 h) into plates containing the compounds, followed by addition of MG (5 µL, 150 µM). After 10 min on ice, the fluorescence was measured at excitation and emission wavelengths of 620 and 665 nm, respectively (EnVision plate reader). Compounds that reconfirmed and did not inhibit MG•MGA fluorescence were tested for concentration-dependent inhibition. For the concentration-response study (n = 2), the concentration of the compounds was varied using two-fold serial dilutions (0.78–100 µM final) and tested following the screening protocol described above, adding the compounds to empty plates using the mosquito X1.
Reconfirmation of Hits
Fresh samples of compounds identified as hits were ordered from commercial vendors for a reconfirmation concentration-response study. This study was done in the same format as the initial concentration-response study, testing the fresh compounds for activity against E. coli βS531L/β′V408G RNAP. The fresh compounds were then tested against a panel of E. coli and MTB RNAPs (10 total, see
Data Analysis
Active compounds from the initial HTS were defined as compounds that decreased fluorescence signal more than or equal to three times the standard deviation of the negative controls (all analyses were calculated on a plate-by-plate basis) and resulted in greater than or equal to 20% fluorescence inhibition. Compounds that the National Institutes of Health does not allow in their screening collection due to reactivity or known toxic substituents (black flags) 22 and compounds with chemical motifs that tend to be highly reactive or highly toxic and nonspecific (red flags) were excluded. Compounds that hit in more than 30% of screens previously run at the CCG were also excluded. Compounds that passed these initial selection criteria were retested (n = 3) and screened for false-positives resulting from inhibition of fluorescence of MGA-bound MG. Compounds that inhibited fluorescence of MGA-bound MG (n = 3) more than 25% were excluded. (Note that two rows on each plate used a less stringent cutoff of 45% fluorescence inhibition because of a partial clog in one tube of the multidrop dispenser cassette, resulting in a lower average fluorescence for these wells.) Remaining compounds with fluorescence inhibition more than or equal to three times the standard deviation of the negative controls in at least two wells (n = 3) were tested for concentration-dependent inhibition. Compounds that inhibited E. coli βS531L/β′V408G RNAP in a concentration-dependent manner (determinable IC50 or displayed concentration-dependent trend) were considered hits from the HTS. Data were fit by nonlinear regression to Eq. (1), where M0 = log of compound concentration, M1 = log of IC50, M2 = hill slope, M3 = lower limit of the curve, and M4 = upper limit of the curve.
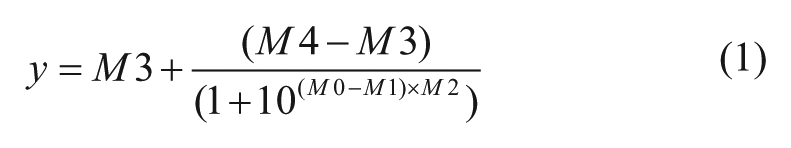
Fluorescence Intercalator Displacement Assay
Binding of compounds to DNA was determined by fluorescence intercalator displacement (FID). A 60 bp FID-lacUV5 probe was used for this study. The FID-lacUV5 probe was hybridized by mixing the top and bottom oligonucleotides (
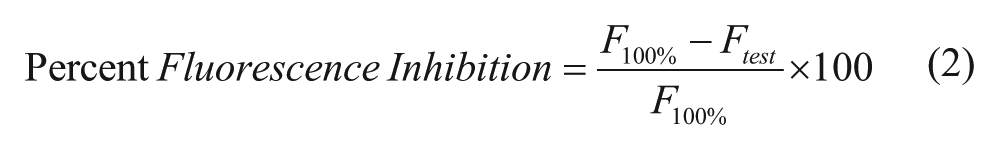
MICs and Cytotoxicity
The MICs of compounds against MTB were determined using MTB H37Rv in a microplate alamar blue assay (MABA) and low-oxygen recovery assay (LORA) as previously described to test the compounds against replicating and nonreplicating bacteria, respectively. 24 The cytotoxicity of the compounds was determined against Vero cells using an MTS assay, as previously described. 25
Results
Development and Validation of Transcription Assay
The pMGA12 plasmid construct was optimized to give a robust signal using an inexpensive detection reagent (MG), so that it was amenable to HTS. To enhance the signal from the initial construct (pTZ18U-4xMGA), several modifications to the plasmid were made (
Hit Identification
A summary of the triage of the HTS is shown in
Figure 2
. The primary screen identified 1738 compounds that met the initial selection criteria (1.2% hit rate). The average Z′ per plate in the primary screen was calculated to be 0.86. The 1738 compounds were then retested in triplicate in the screening assay and also for direct inhibition of MG•MGA fluorescence. Of the 1738 compounds, 396 compounds replicated the screening assay results in triplicate (~25% reconfirmation); however, 89 of these compounds were removed from consideration because they directly inhibited the MG•MGA fluorescence. This yielded 307 compounds, which were tested further. The confirmation and control screening identified 234 of the 307 compounds that displayed concentration-dependent inhibition of transcription, with 190 compounds having a determinable IC50 (<100 µM, the highest concentration tested;

Flow chart of selection criteria for high-throughput screen. Values in parentheses are the number, or percentage, of compounds that did not pass that particular selection criteria. *As discussed in the Data Analysis section, a less stringent cutoff (45%) was used for two rows of the 384-well plates in the MG•MGA control screen.
Reconfirmation
Fresh samples of the 63 compounds were purchased and retested for activity. Of the 63 compounds, 30 compounds were inactive against E. coli βS531L/β′V408G RNAP; however, 27 of the inactive compounds are members of structural clusters, which contain active analogs. To retain chemical diversity within the scaffold clusters, we elected to eliminate only the three inactive compounds that were not in such clusters. This yielded 60 compounds, which were then tested for activity against a panel of E. coli and the corresponding MTB RNAPs: WT, three different RifR mutants (βD516V, βH526Y, βS531L; E. coli numbering) as well as the double mutant with a compensatory mutation (βS531L/β′V408G), which was the target of the HTS. The IC50 values of the compounds against these RNAPs ranged from 5 µM to >200 µM, with the majority of the compounds having similar values across the RNAP enzymes tested. The IC50 values for the most active compounds against each enzyme are in
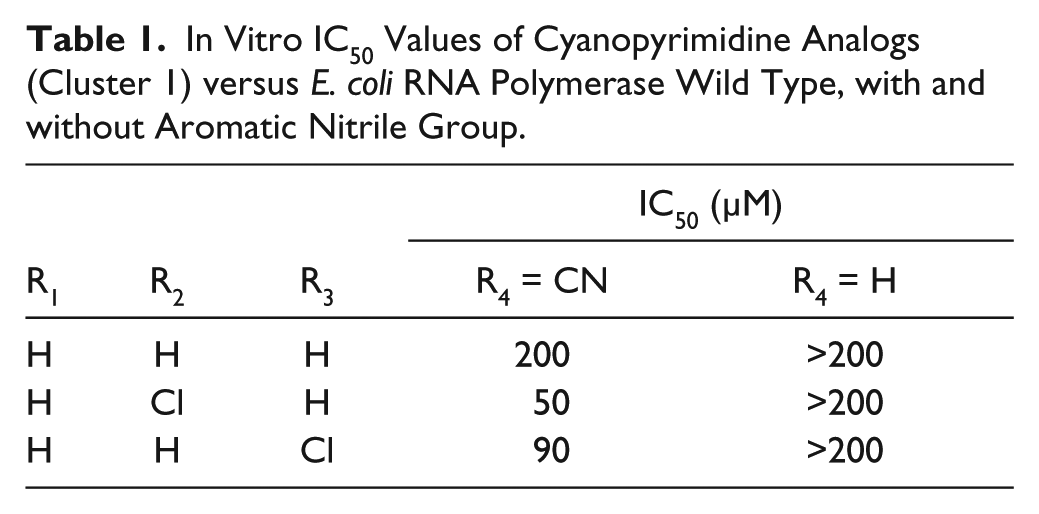
In Vitro IC50 Values of Cyanopyrimidine Analogs (Cluster 1) versus E. coli RNA Polymerase Wild Type, with and without Aromatic Nitrile Group.
MICs and Cytotoxicity
Compounds with IC50 values less than 50 µM against at least one RNAP (18 compounds) were tested for inhibition of replicating (MABA) and nonreplicating (LORA) MTB. Of the compounds tested, only four compounds exhibited detectable growth inhibition at concentrations lower than 200 µM (
Table 2
and
MICs of Active Compounds versus M. tuberculosis H37Rv.
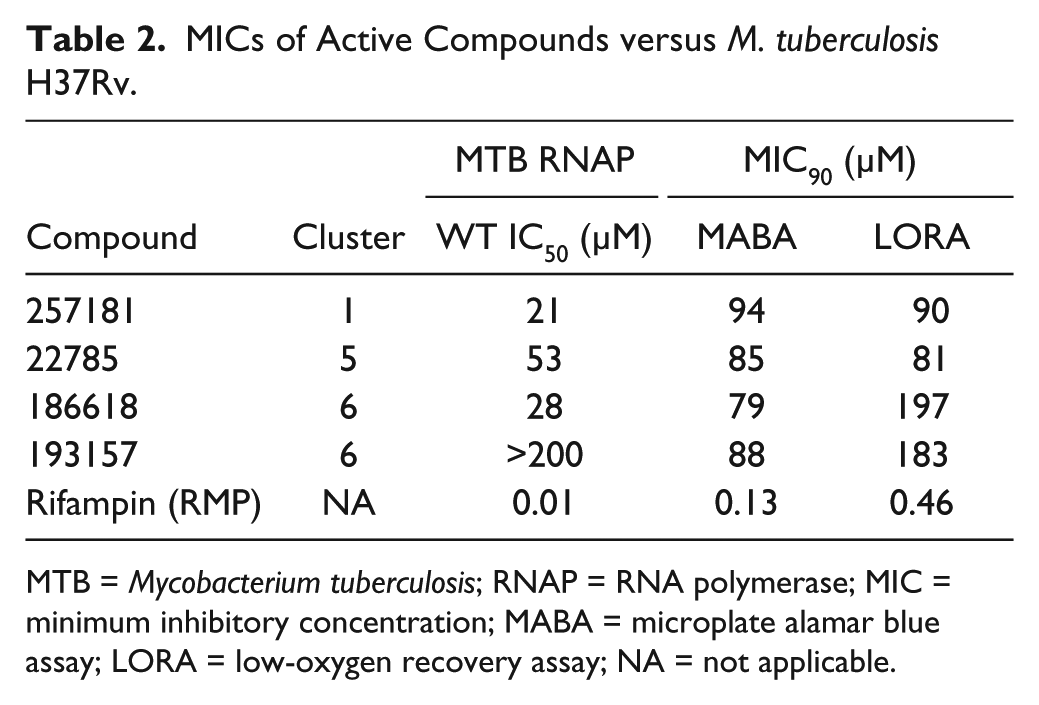
MTB = Mycobacterium tuberculosis; RNAP = RNA polymerase; MIC = minimum inhibitory concentration; MABA = microplate alamar blue assay; LORA = low-oxygen recovery assay; NA = not applicable.
FID Assay
Compounds active against E. coli or MTB RNAP in vitro were each tested at four different concentrations (12.5–100 µM) using the FID assay to determine if the compounds potentially inhibit RNAP by binding DNA. Berenil (diminazine aceturate), a known minor groove binder, was used as a control for assay validation.
26
Three of the compounds inhibited fluorescence of ethidium bromide in the FID assay at 100 µM (one of them had some inhibition at 50 µM also) using the FID-lacUV5 probe (
Discussion
MTB RNAP is a well-validated target for the treatment of TB; however, target-based mutations conferring resistance to RMP have resulted in a large number of RifR strains in clinical isolates. The majority (~96%) of RifR isolates have site-specific mutations in a 27 amino acid region of RNAP where RMP binds (RRDR). The potent sterilizing activity (i.e., bactericidal against both active and latent MTB) of RMP against WT MTB indicates that RNAP is still a promising target for the treatment of RifR MTB if the rifamycin (Rif) resistance can be circumvented. Previous assays to identify novel inhibitors of bacterial RNAP have used nonnative substrates, which can be cost prohibitive for HTS.11,12 An assay amenable to HTS using the commercially available RiboGreen has been reported; however, in addition to the cost associated with the RiboGreen dye, it requires removal and purification of RNA from the DNA template since RiboGreen fluorescence is enhanced by binding to DNA as well as RNA. 16 An assay has been developed that monitors the increase in fluorescence of MG upon binding to an RNA aptamer specific for triphenylmethane dyes. 27 The MGA has been applied to detection of E. coli RNAP activity via rolling circle transcription of a small, circular, single-stranded DNA template. 15 We adapted this approach to use the aptamer for detection of RNA transcribed from a double-stranded DNA template. Obtaining quantities of MTB RNAP sufficient for an HTS has been prohibitive. However, the E. coli RNAP can be obtained in sufficient quantities, has greater activity, and is more stable than the MTB RNAP. We have shown that the E. coli RNAP is a reasonable model for the MTB RNAP, 16 and we have solved the X-ray crystal structure of the E. coli RNAP alone and in complex with Rif derivatives that we have designed. 21 This provides us the potential to determine the three-dimensional structure of the RNAP complex with any novel RNAP inhibitor that we find that has sufficient affinity.
An HTS of 150,554 small molecules was then carried out against E. coli RNAP βS531L/β′V408G ( Fig. 1B ) using the aforementioned plasmid-based in vitro transcription assay. As discussed in the Results section and shown in Figure 2 , the concentration response, control screening, and triage for potential toxicity, novelty, redundancy, and commercial availability resulted in the selection of 63 compounds for reordering. The fresh samples were screened against a panel of E. coli and the corresponding MTB RNAPs: wild-type, three different RifR mutants (D516V, H526Y, S531L; E. coli numbering), and the double mutant (βS531L/β′V408G). The IC50 values of the compounds against these RNAPs ranged from 5 to >200 µM (data not shown). With the exception of cluster 6, discussed below, we selected scaffolds that do not show any large (>10-fold) differential in IC50 values across the panel of WT and RifR RNAP mutants tested, as these would seem to be more promising as they are likely to be binding outside of the RRDR, hence lacking cross-resistance with RifR RNAPs.
Encouragingly, 9 of the 63 compounds ( Table 3 ) had similar IC50 values across the panel of RNAP enzymes tested, indicating that they are active against RifR mutants. However, only two of these compounds were active against MTB in culture (257181 and 22785). Compound 257181 belongs to a group of compounds (cluster 1, cyanopyrimidines) identified with similar IC50 values in vitro. Two of the compounds in cluster 1 (257180 and 257182) inhibited fluorescence in the FID assay, indicative of DNA binding; however, this occurred only at concentrations >10-fold higher than their RNAP IC50s. Therefore, it is reasonable to deduce that these compounds do not exhibit their RNAP inhibitory activities via DNA binding. Initially, this cluster of compounds was attractive for further development. However, the cyanopyrimidine scaffold was identified by a chemoinformatic filter that identifies compounds that are likely to exhibit pan-assay interference (PAINS; A. White, unpublished data) Further, compounds with aromatic nitriles have been reported to form covalent bonds with cysteines. 28 To determine if this scaffold is indeed a false-positive because of PAINS, a set of six analogs of cluster 1, with and without a nitrile at position R4, were purchased, and IC50 values were determined with WT E. coli RNAP. All of the analogs tested that lacked a nitrile group on the pyrimidine ring were inactive ( Table 1 ). Based on these results, it was concluded that this scaffold was not worth pursuing further.
Compounds with Similar Activity across WT and RifR RNA Polymerase.
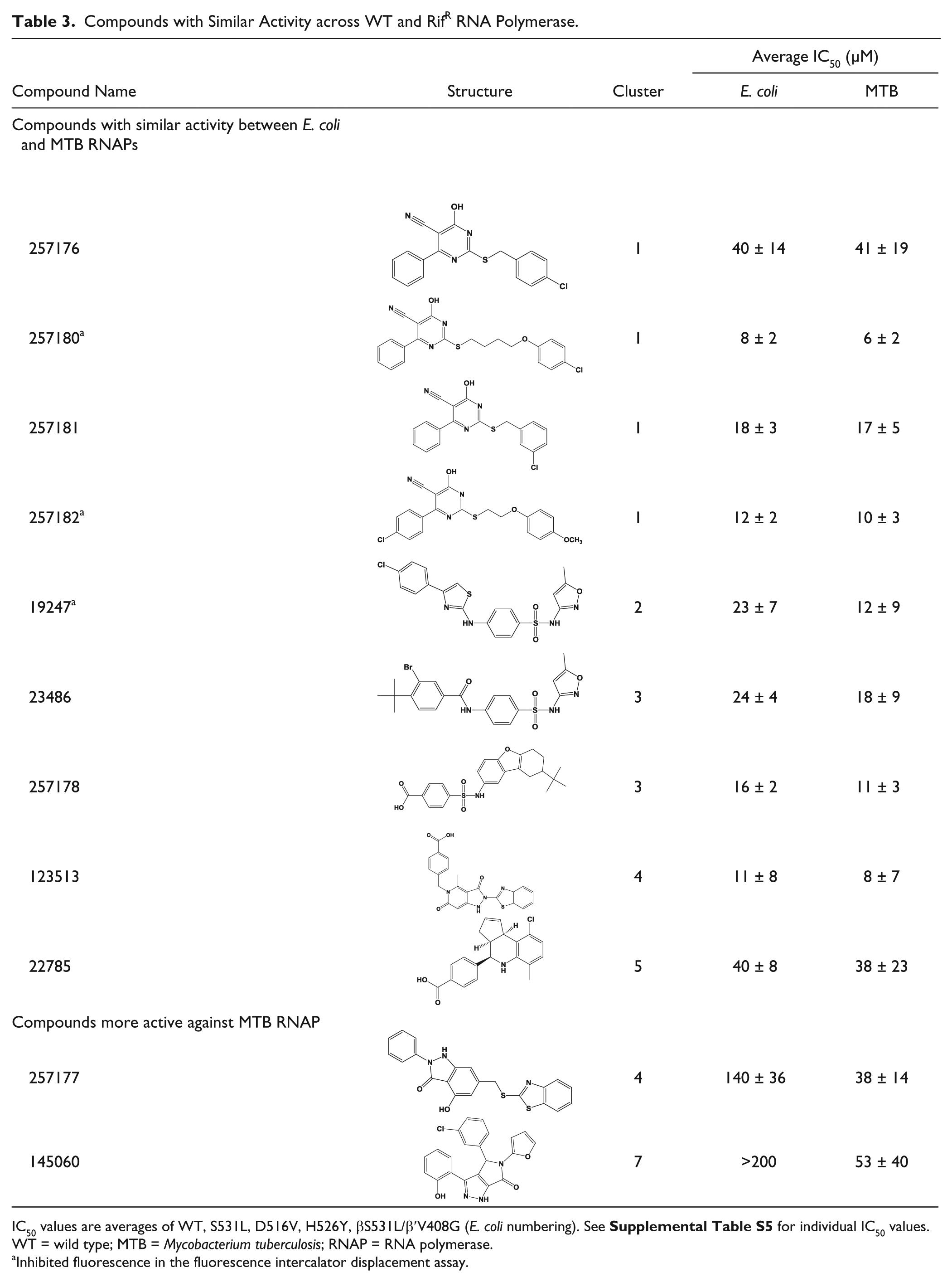
IC50 values are averages of WT, S531L, D516V, H526Y, βS531L/β′V408G (E. coli numbering). See
Inhibited fluorescence in the fluorescence intercalator displacement assay.
Another group of compounds ( Table 4 , cluster 6, pyrazolopyrimidines) showed comparable activity across most of the E. coli RNAPs with the exception of H526Y. A recent crystal structure of E. coli H526Y RNAP shows significant conformational changes in the Rif binding pocket relative to the WT, D516V, and S531L RNAPs (V. Molodtsov et al., unpublished data) This suggests that these compounds may bind in the Rif binding pocket, but their much smaller size relative to RMP allows them to bind to the D516V and S531L RifR mutants. As precedence, Xu et al. 29 showed that E. coli H526Y RNAP had complete resistance to the RNAP inhibitor sorangicin A while mutants D516N and S531F, and other residues, showed only partial or no resistance.
IC50 Values (µM) against E. coli and MTB RNAPs for Compounds with Reduced Activity against E. coli H526Y RNAP (Cluster 6).
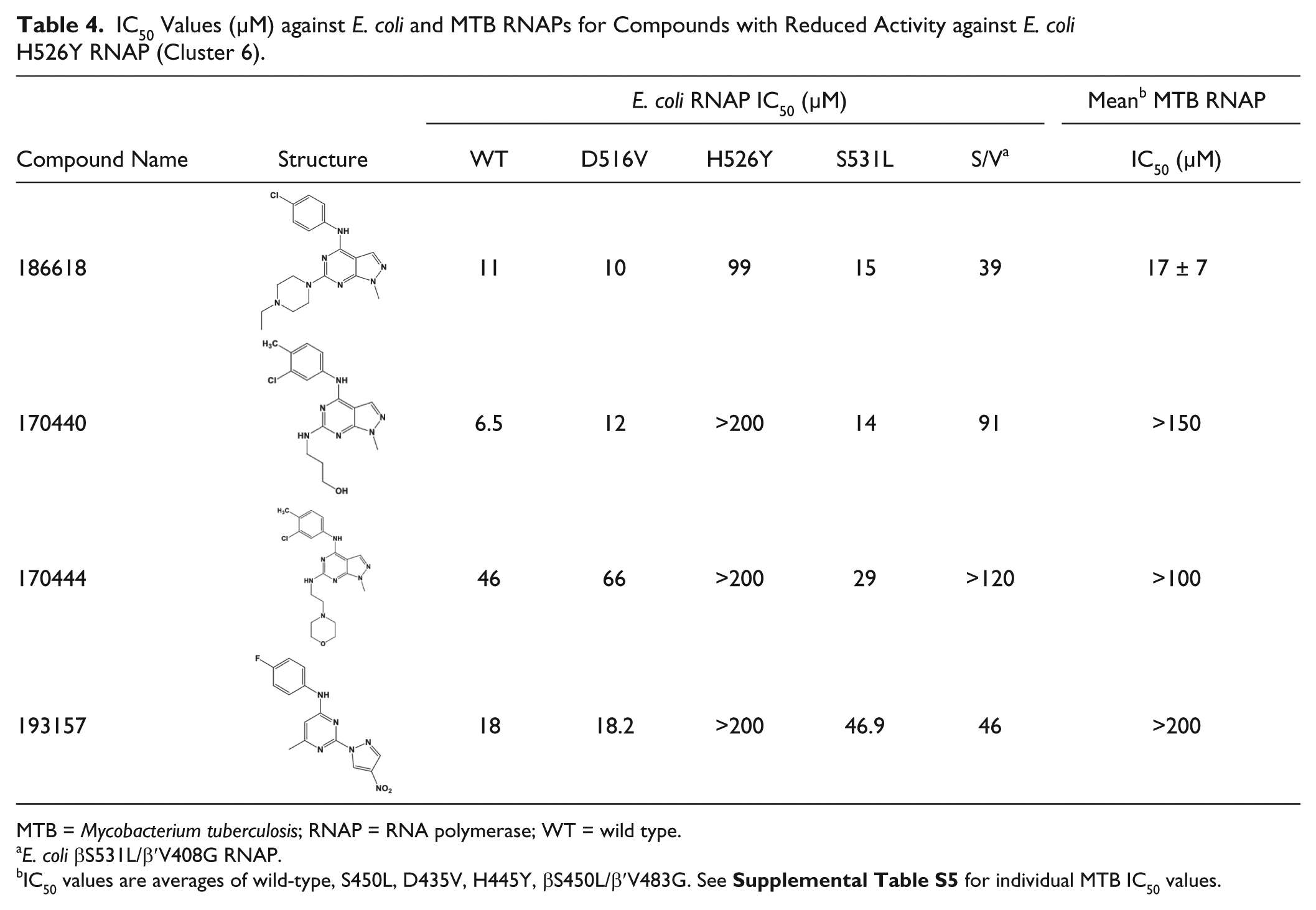
MTB = Mycobacterium tuberculosis; RNAP = RNA polymerase; WT = wild type.
E. coli βS531L/β′V408G RNAP.
IC50 values are averages of wild-type, S450L, D435V, H445Y, βS450L/β′V483G. See
Two of the four compounds in cluster 6 (186618 and 193157) showed activity against MTB in culture ( Table 2 ). Interestingly, compound 193157 was active in culture despite being inactive against WT MTB RNAP in vitro. It is possible that the activity of 193157 against MTB is due to off-target or nonspecific activity. However, it is known that MTB does metabolize a number of drugs to more active forms30,31; therefore, our compound may be metabolized (e.g., reduction of the nitro) to an active, RNAP-inhibiting derivative. Compound 186618 was active against all RNAP enzymes tested and active against replicating and nonreplicating MTB. This compound has been synthesized, and the structure and purity were confirmed (R. Rowlands and A. White, unpublished) and found to exhibit very low activity, similar to other cluster 6 analogs. We suspect that the purchased sample of 186618 was contaminated with something that was responsible for the activity. This is under investigation.
Two compounds that have higher activity against MTB RNAPs relative to E. coli RNAP ( Table 3 ) were identified. Compound 257177 is in the same cluster as 123513 ( Table 3 , cluster 4) but has a significantly different activity profile. Compound 257177 is ~10-fold less active against E. coli RNAP than 123513. We cannot make any significant correlations between the structures and activities of these two compounds; however, these differences are noteworthy for potential future structure activity relationship studies in optimizing activity for MTB (or E. coli) RNAP. Compound 145060 showed no significant activity against E. coli RNAP in the follow-up assays and only modest activity against MTB RNAPs (>50 µM).
In this report, we have described an efficient method for detecting transcription of bacterial RNAP from a plasmid template that is suitable for HTS (Z′ = 0.86). Using this novel assay, we identified a set of 15 compounds from seven different structural scaffolds that exhibited promising activity against E. coli and/or MTB RNAP for further investigation. Chemoinformatic analysis and follow-up experiments allowed us to eliminate one scaffold due to likely pan assay interference (PAINS). Ten of the 15 compounds (see
Footnotes
Acknowledgements
We would like to acknowledge our colleagues at the University of Michigan: Dr. Kristen Wiese for construction of pTZ18U-4xMGA, Dr. Irosha Nawarathne for construction of pVS10 (S531L/V408G), Max Stefan for construction of pACYCDuet-rpoA-rpoZ, pETDuet-rpoB-rpoC (WT, S450L, S450L/V483G) and pRSF-SigA, Martha Larsen and Dr. Nicholas Santoro at the U-M Center for Chemical Genomics for help with HTS, Rachel Rowlands and Dr. Andrew White for the synthesis of 186618, and Drs. Hollis Showalter, Andrew White, and Paul Kirchhoff for help with the HTS triage and scaffold selection. We would also like to acknowledge Dr. Baojie Wan and Professor Scott Franzblau of the University of Illinois, Chicago Institute for Tuberculosis Research, for the MIC determinations. We would also like to acknowledge members of the Garcia lab for critical reading and helpful suggestions about the manuscript.
Supplementary material is available online with this article.
Author Contributions
N.T.S. and G.A.G. conceived and designed the experiments. K.S.M. conceived, designed, and oversaw enzyme purification for HTS. V.M. purified the enzyme used for the high-throughput screen, and N.T.S. purified enzymes for reconfirmation studies. N.T.S. performed the experiments. N.T.S. and A.K. constructed the pMGA12 DNA template. N.T.S. and G.A.G. wrote the initial draft and revision of the manuscript, and all authors discussed the results and edited the initial manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institutes of Health grants R01AI110780-01 (G.A.G.) and T32GM007767 (NTS trainee), the University of Michigan College of Pharmacy, and the University of Michigan Center for the Discovery of New Medicines.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.