
Abstract
New and improved drugs against tuberculosis are urgently needed as multi-drug-resistant forms of the disease become more prevalent. Mycobacterium tuberculosis cytidylate kinase is an attractive target for screening due to its essentiality and different substrate specificity to the human orthologue. However, we selected the Mycobacterium smegmatis cytidylate kinase for screening because of the availability of high-resolution X-ray crystallographic data defining its structure and the high likelihood of active site structural similarity to the M. tuberculosis orthologue. We report the development and implementation of a high-throughput luciferase-based activity assay and screening of 19,920 compounds derived from small-molecule libraries and an in silico screen predicting likely inhibitors of the cytidylate kinase enzyme. Hit validation included a counterscreen for luciferase inhibitors that would result in false positives in the initial screen. Results of this counterscreen ruled out all of the putative cytidylate kinase inhibitors identified in the initial screening, leaving no compounds as candidates for drug development. Although a negative result, this study indicates that this important drug target may in fact be undruggable and serve as a warning for future investigations.
Introduction
In 2013, 1.5 million people died from tuberculosis (TB), making it the second leading cause of death due to a single infectious organism in the world, behind HIV/AIDS. 1 Also that year, multi-drug-resistant TB (MDR-TB) was estimated to have developed in 480,000 people, 1 highlighting the emergence of MDR-TB and the need for new more potent drugs against the disease.
Novel drugs against TB can be identified by screening molecular libraries for inhibition of potential protein targets. One such pool of potential targets includes proteins that have been structurally characterized by high-resolution X-ray crystallography. Since these proteins have already been expressed, purified, and crystalized, lead hit compounds may be co-crystalized with the target protein providing vital information for the design of compound variants with increased selectivity, binding efficiency, and potency.
Unfortunately, many of the proteins that would be good candidates for screening have not been structurally characterized because they fail somewhere along the gene-to-structure pipeline. Indeed, this applies to approximately 90% of the Mycobacterium tuberculosis (Mtb) proteomes attempted and is a major barrier to drug development. 2 While genetic engineering of problematic targets to introduce truncations, point mutations, and alternative affinity tags can rescue the target for structure determination, this process is labor-intensive and not conducive for implementation in a high-throughput (HT) screening format. An alternative approach is to use homologue proteins from closely related species, that is, orthologues, which may have different solubility and characterization properties from the TB protein, yet retain the same function and have high sequence similarity at the active site.
A recent analysis of the TB proteome has shown that it is possible to simply and quickly predict high active site similarity based on a threshold of 55% global sequence similarity between a TB and non-TB orthologue pair. 2 Of 106 protein pairs analyzed, it was shown that 90% of those with >55% global sequence identity had a highly similar active site based on three criteria. The first criterion was a measure of the average distance between common Cα atoms in two aligned structures (Cα root mean square deviation [RMSD]) at the active site of <1 Å. The second was an active site side-chain identity of >85%, and the third criterion was a measure of active site shape and chemical similarity using seven pharmacophoric (chemical) properties derived from structural data of >80%. Combined, these three criteria indicate highly conserved active site shape and chemistry between the two orthologues of the pair.
One such orthologue pair is for cytidylate kinase (CK), which plays an important role in the synthesis and salvage pathways of DNA and RNA precursors.3,4 The Mtb enzyme is essential in vitro,5,6 and differs in its substrate specificity from the human orthologue, uridine/cytidine monophosphate kinase,7,8 making it an ideal drug target. No structure data exist for the Mtb enzyme; however, structures were solved for the homologue from Mycobacterium smegmatis (Msm) (PDB ID: 3R20) and from Mycobacterium abscessus (PDB ID: 3R8C). These proteins have 68% and 74% global sequence identity to the Mtb enzyme and 73% to each other. Furthermore, their active sites were shown to have nearly identical side-chain orientations surrounding the substrate. 2 It seems very likely then that the active site will also be highly conserved in the Mtb structure, and that either the Msm or the Mab protein can be used as a surrogate in developing drugs against the Mtb enzyme.
Here we report on a luciferase-based assay for Msm CK activity and HT screening of 20,000+ compounds.
Materials and Methods
Cloning, Expression, and Purification
PCR amplification, cloning, sequencing, expression screening, scale-up, and purification of the Msm CK protein were performed as described previously for the Seattle Structural Genomics Center for Infectious Disease (SSGCID) gene-to-structure pipeline targets. 9
High-Throughput Activity Assay
CK catalyzes the reaction ATP + (d)CMP ADP + (d)CDP. Activity was determined by the measurement of ATP accumulated in the reaction ADP + (d)CDP + enzyme = ATP + (d)CMP. ATP generated was measured by the luciferase-based Kinase-glo luminescent assay platform (Promega, Madison, WI). Briefly, our protocol was as follows: On ice, 5 µL assay buffer (50 mM Tris, 50 mM KCl, 2 mM MgCl2, 0.5 % NPS, pH 7.5) was dispensed to each well of a 384-well assay plate. Then 5 µL of a 2× master mix of 4 µM ADP, 16 µM CDP, and 300 ng/µL purified CK protein in assay buffer was added to each well. Each plate was covered and incubated at 37 °C for 110 min. Plates were removed from 37 °C and equilibrated to room temperature. Ten microliters of Kinase-glo reagent was added to each well, and then the plates were incubated at room temperature for 10 min. Luminescence was measured at 525 nm on an EnVision MultiLabel Reader (PerkinElmer, Waltham, MA). Since no suitable inhibitors for CK were reported in the literature, background signal was determined by dropping CDP from the reaction.
This protocol was adapted for HT screening at the Fred Hutchinson Cancer Research Center (Fred Hutch, Seattle, WA) in the following manner: After dispensing 5 µL of assay buffer to each assay plate well, 0.06 µL of compound dissolved in 100% DMSO was pin-transferred from the compound library plate to a final concentration of approximately 12 µM. Master mix was then dispensed to each well with a Microflo Select Dispenser (BioTek Instruments, Winooski, VT). The assay plates were then treated as previously described. Compounds were tested in duplicate, and each plate had one column of background only and one column of no-drug experimental control wells.
Data Processing
Prior to screening, compound mapping files containing plate coordinates and barcode identifiers were uploaded to HutchBASE, a custom laboratory information management system (LIMS). A Biomek FX liquid handling system (Beckman Coulter, Inc., Brea, CA) configured with a FP4 magnetic-mounted 384-well pinning tool (S&P Robotics, Inc., Toronto, ON), a barcode scanner, and sample tracking software were used to record liquid transfer processes. The EnVision Multi-Label Reader (PerkinElmer) was configured with a barcode scanner and recorded luminescence measurements employing the ultrasensitive luminescence mode. Raw data files were uploaded to HutchBASE for further processing and analysis using the Bioconductor package plugin cellHTS2. 10 Data were log2 transformed, normalized to the plate median, standardized, and scored using a robust Z-score method. For each assay replicate the overall median and the median absolute deviation (MAD) were used in place of the mean and standard deviation, respectively. Z-score results from experimental replicates were averaged for final reporting.
A total of 17,300 compounds from two distinct libraries were screened at the Fred Hutch facility in this manner ( Table 1 ).
Summary of HT Screening Results from Fred Hutch Facility.

Hit Validation
Hit compounds were selected based on a Z score of −5 or below. Percent inhibition for each hit compound was calculated using the duplicate average absorbance reading at 525 nm (A525) for each compound well and the column average A525 reading for the no-drug control wells of that plate. Each hit compound was repicked from the master drug plate and assayed again. Compounds that retested at less than 7.5% inhibition were eliminated. Additionally, to identify false positives due to the inhibition of luciferase activity in the Kinase-glo solution, a counterassay for compounds that decreased luminescence was performed. Compounds that inhibited the signal from a 5 µM ATP solution by more than 15% were deemed false positives or insufficiently selective for further study. As a further validation, compounds that still showed inhibition of CK and little to no inhibition of luciferase were analyzed for dose response to the compound from 1 to 240 µM in both the CK assay and the luciferase counterassay.
Mycobacterium Whole Cell Active Compound Screening
The CK activity assay was also adapted to HT screening at Texas A&M University. The assay was performed in a similar fashion but differed in its utilization of a CyBi-well Vario robotic pipetting station (CyBio, Jena, Germany) combined with a Polarstar Omega plate reader (BMG Labtech, Ortenberg, Germany). Approximately 2600 compounds that previously were identified as inhibitory to M. tuberculosis H37Rv growth 11 were obtained from the Tuberculosis Antimicrobial Acquisition and Coordinating Facility at the Southern Research Institute and were screened as singlets at a concentration of approximately 20 μM.
Thermal Melt Assay
A thermal shift assay, which detects ligand binding to protein as an increase in the protein melting temperature, was performed as described previously 12 with the leading compounds from HT screening. Purified CK at 300 ng/µL and 120 µM compound in assay buffer with SYPRO Orange fluorescent dye (Sigma, St. Louis, MO) were heated from 20 °C to 90 °C in 96-well plates more than 55 min. Dye incorporated into the protein was measured at 575 nm luminescence, and melting temperatures were derived from the resultant melt curves.
Computational Analysis of Novel Drug Opportunities Platform
The Computational Analysis of Novel Drug Opportunities (CANDO) platform uses similarity of drug–proteome interaction signatures to infer homology of drug behavior (http://protinfo.org/cando). For the first version of the platform, we constructed interaction signatures between 3733 human-ingestible compounds mapping to 2030 indications and 48,278 protein structures based on basic science methodologies to predict and analyze protein structure, function, and interactions developed by us and others.13,14 Our signature comparison and ranking approach yielded benchmarking accuracies of 12%−25% for 1439 indications with at least two approved compounds. We prospectively validated 58/163 (35%) “high-value’’ predictions from 12 in vitro studies covering 10 indications, with comparable or better activity than existing drugs, which serve as novel repurposed therapeutics.
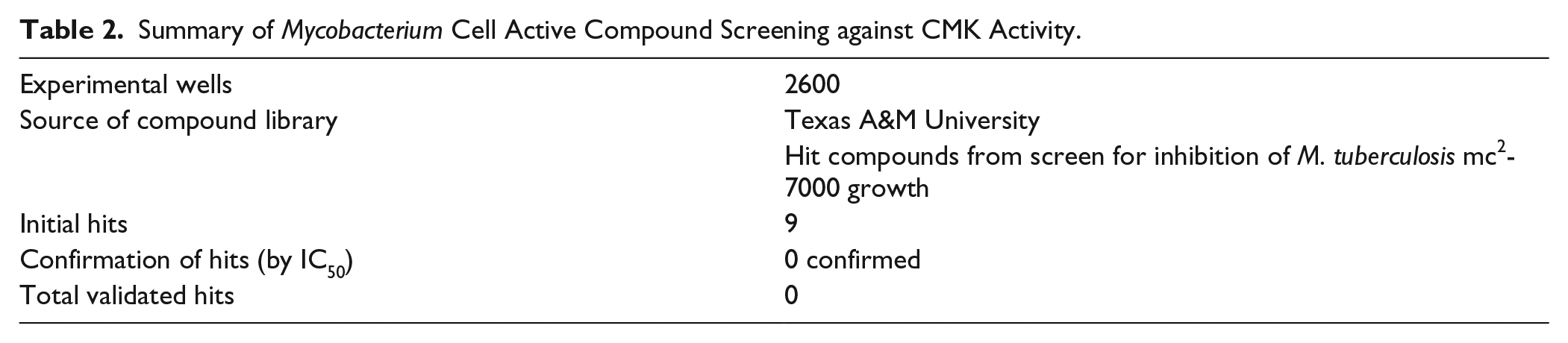
CANDO represents a proteomic approach to drug discovery, and the bioinformatic docking algorithms were not optimized to work for single targets. Nonetheless, since we had applied the CANDO v1 platform against the entire M. tuberculosis proteome, the interaction scores between the 3733 compound library and CK were readily available. We extracted this information, sorted the scores, and identified the top 20 compounds that were most likely to interact with CK. These compounds were tested in duplicate for inhibition at low and high concentrations ( Table 2 ) in the same format as the hit validation assays.
Summary of Mycobacterium Cell Active Compound Screening against CMK Activity.
Results and Discussion
HT Screening and Validation
A total of 17,300 compounds were screened in duplicate from two compound libraries at Fred Hutch ( Table 1 ). Z′ values were calculated for each plate and all were above 0.7, indicating sufficient separation between positive and negative controls ( Fig. 1 ). Likewise, signal to noise (S/N) ratios calculated for each plate using the no-drug and background well A525 readings fell within an acceptable range (S/N: 8.8–17.0). Additionally, edge effects were observed to be minimal. A total of 22 selected compounds had Z scores of less than −5 and were further assayed for validation.
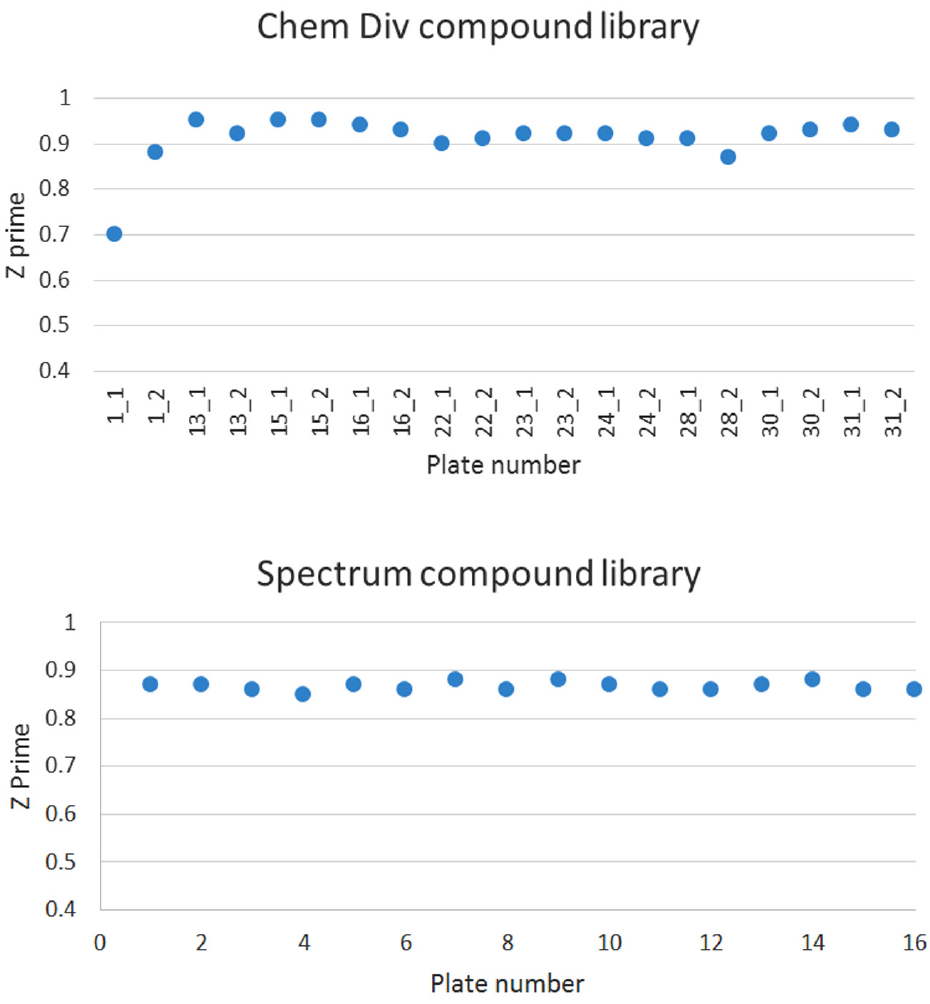
Z′ values per hit-containing plate for HT screening at Fred Hutch.
Of the 22 compounds retested, 16 showed repeated inhibition of CK >7.5% ( Table 1 ). We chose 7.5% inhibition as the cutoff since it correlated with the −5 Z-score cutoff for selecting statistically significant hits in the HT screen. However, inhibitors of the ATP-dependent luciferase generation of light could be false positives in this screen. Indeed, of those 16 compounds, 10 were eliminated from further study because the luciferase counterscreen suggested they were luciferase inhibitors and not likely CK inhibitors. Three of the remaining six compounds were also eliminated because they inhibited the CK reaction weakly (7.5%−15% inhibition) and they also showed similar weak inhibition of the luciferase (10%−15%). Had these compounds been strong CK inhibitors or showed 0% inhibition of luciferase, their inclusion in the next round of validation could have been justified.
The remaining three compounds were repurchased and tested against CK and luciferase in IC50 experiments. The CK IC50 for duplicate wells ranged from 3 to 120 µM, reconfirming the compounds’ inhibition in the CK assay ( Table 1 ). However, all three of the compounds had luciferase IC50 results of less than 3–120 µM ( Table 1 ). These results are in contrast to the 0% luciferase inhibition observed in the initial counterassay and suggest these were false-positive inhibitors of luciferase, not detected in the original luciferase counterassay. Of the three compounds, indoprofen is a known luciferase inhibitor with an IC50 of greater than 50 µM reported for the same Kinase-glo luciferase (Promega) used in this assay. 15 The compound phenylmercuric acetate (PMA) has an even lower luciferase IC50 of 3 µM and was also ruled out. The remaining compound, ChemDiv C202-3495, had weak luciferase inhibition with an IC50 of 120–240 µM ( Table 1 ), so it was further analyzed by testing in a thermal shift assay for the CK enzyme. No thermal shift of the enzyme with the compound was observed, while a shift of approximately 3 °C was observed for the control substrate CDP at 5mM. Since we could not demonstrate physical association with the CK enzyme, it seemed likely that this compound was also acting through inhibiting the luciferase reaction itself. Thus, the HT screen of >17,000 diverse compounds failed to demonstrate a true CK inhibitor.
Mycobacterium Cell Active Compound Screening
We also screened a battery of 2600 compounds that were active against M. tuberculosis, in case any of these compounds found as phenotypic hits could be shown to inhibit CK. Nine compounds were observed as having some inhibitory effect on CK in the initial screening test and were chosen for retest. Serial dilution series of the compounds from 50 µM to 10 nM were prepared and tested in the assay. None of the compounds demonstrated a dose-dependent response, and none of the phenotypic hits appear to inhibit CK ( Table 2 ).
Screening of CANDO Compounds
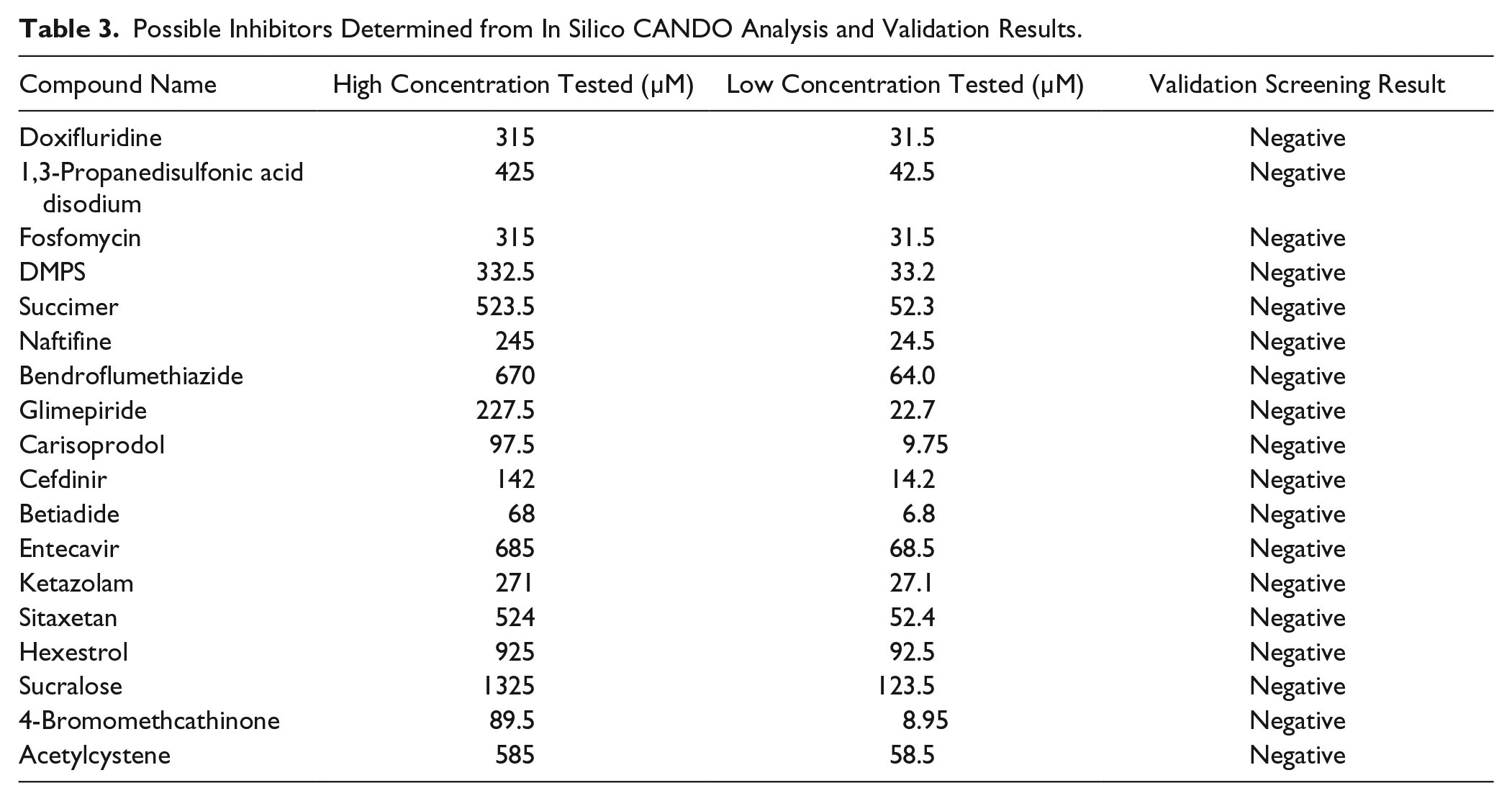
Eighteen of the top-ranked compounds predicted to be top inhibitors of CK from an in silico docking screen using the CANDO software were purchased and tested in duplicate in the CK assay ( Table 3 ). No inhibition was observed for any of these compounds, even at concentrations as high as 1.3 mM. Although this particular result is a negative one, it suggests that this pioneered multitarget approach may be more effective against M. tuberculosis.13,16,17 CANDO works in part because docking algorithms are not perfect when considered on a target-by-target basis, but on average, across entire proteomes, they are more accurate.13,14 Whereas CANDO may work in the aggregate at treating a disease by inhibiting a pathogen, this may not necessarily be the case for particular single targets of interest.
Possible Inhibitors Determined from In Silico CANDO Analysis and Validation Results.
Prospects for Drug Development
In addition to the 19,900 compounds reported here, we tested CK against staurosporine, a potent protein kinase inhibitor, to assess the potential of screening against protein kinase inhibitor libraries. There was no inhibition observed with staurosporine; thus, we chose not to pursue libraries enriched for protein kinase inhibitors.
Given the zero hit rate from the HT and CANDO validation screening, additional screening for inhibitors does not seem fruitful for CK. However, we feel it is important to publish when a HT screen has been performed with a promising drug target like CK, no matter the result. Indeed, publications have hypothesized that CK would be a good drug target.2,18 While we cannot exclude that alternate approaches such as screening with natural product libraries may yet yield results, we feel that this publication can serve as a warning that CK may be undruggable.
Footnotes
Acknowledgements
Steve Nakazawa-Hewitt, Ryan Choi, Janette Myers, and our other UW-PPG staff within the Seattle Structural Genomics Center for Infectious Disease (SSGCID) performed cloning, expression, and purification of the Msm CK protein. Greg Crowther provided advice on assay development. Peter J. Myler, Robin Stacey, and the SSGCID Scientific Leadership Team (SLT) provided general guidance for the project.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported with federal funds from the National Institute of Allergy and Infectious Diseases Contract HHSN272201200025C, entitled “Structural Genomics Centers for Infectious Diseases.”
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.