
Abstract
The prevailing hypothesis of ketamine’s unique antidepressant effects implicates N-methyl-d-aspartate receptor (NMDAR) inhibition-dependent enhancement of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor-mediated transmission, activation of intracellular signalling pathways and increased synaptogenesis. Recently, however, a seminal study by Zanos et al. directly challenged the NMDAR hypothesis of ketamine with the claim that an active ketamine metabolite, (2R,6R)-hydroxynorketamine, devoid of NMDAR binding properties or key side effects of its parent compound, is both necessary and sufficient for ketamine’s antidepressant effects in rodents. However, following these encouraging initial findings, one preclinical study failed to replicate the antidepressant effects of (2R,6R)-hydroxynorketamine (HNK), while others have questioned the metabolite’s contribution to ketamine’s therapeutic effects or argued against rejecting the NMDAR hypothesis of ketamine action. In light of these potentially paradigm-shifting, but highly controversial, findings, this review will summarise and critically evaluate the evidence for and against the NMDA receptor hypothesis of ketamine action, with a particular focus on (2R,6R)-HNK and the implications of its discovery for understanding ketamine’s mechanism of action in depression. Ultimately, uncovering the molecular mechanisms underlying the therapeutic effects of ketamine and possibly (2R,6R)-HNK, will aid the development of novel and more efficacious antidepressant agents so urgently needed to address a major public health concern, and could hold potential for the treatment of other stress-related psychopathologies, including bipolar disorder, post-traumatic stress disorder and suicidality.
Keywords
Introduction
Current antidepressants have important shortcomings, particularly the relatively low clinical response rates and delayed onset of action, highlighting a clear unmet clinical need for novel, superior antidepressants.1,2 One of the most exciting discoveries in the field of psychiatry in the last two decades is that a single sub-anaesthetic dose of ketamine, the non-competitive, open-channel N-methyl-d-aspartate receptor (NMDAR) antagonist, has robust, rapid and sustained antidepressant effects in individuals with major depressive disorder (MDD) or bipolar disorder.3–7 Clinical trials demonstrate that a single 40-min-long intravenous (IV) infusion of (R,S)-ketamine (0.5 mg/kg) results in response rates of 50%–70% in treatment-resistant depression (TRD – failed two or more first-line antidepressants).1,7,8 In stark contrast to classical antidepressants, ketamine possesses rapid antidepressant effects that are evident within an hour, peak around 24 h and can last up to 1–2 weeks after infusion.7,9 Unfortunately, the widespread clinical use of ketamine for depression is limited by psychotomimetic side effects, abuse potential and potential neurotoxicity with repeated exposure.3,10
These encouraging clinical findings have inspired preclinical research aimed at understanding how NMDA receptor inhibition mediates ketamine’s clinical efficacy, as well as, tremendous interest in alternative NMDAR antagonists as novel antidepressants.6,11–13 Although ketamine’s therapeutic effects were long attributed to direct NMDAR inhibition, emerging clinical and pre-clinical data cast significant doubt on the oversimplified NMDAR inhibition hypothesis of ketamine’s action in depression. 13 Most recently, a seminal Nature paper by Zanos et al. 14 directly challenged the NMDAR hypothesis of ketamine by claiming that an active metabolite, (2R,6R)-hydroxynorketamine (HNK), is both necessary and sufficient for ketamine’s antidepressant effects in rodents, and importantly, does not inhibit the NMDAR. Moreover, (2R,6R)-HNK lacked key side effects of the parent compound, including sensory-dissociation effects and abuse potential in mice. 14 Thus, this study suggests that ketamine’s therapeutic effects are not mediated by its ability to block NMDARs as previously assumed, but require ketamine’s metabolism into (2R,6R)-HNK, which achieves its primary antidepressant activity by enhancing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) function. 14 Subsequent research and commentaries question the original findings and urge caution in rejecting the NMDAR hypothesis of ketamine action.15–18 The most telling finding is the failure to replicate (2R,6R)-HNK’s antidepressant effects in two different tests of depression. 17 In light of these potentially paradigm-shifting, but highly controversial, findings, this review will summarise and critically evaluate the evidence for and against the NMDA receptor hypothesis of ketamine action, with a particular focus on (2R,6R)-HNK and the implications of its discovery on our understanding of ketamine’s mechanism of action in depression.
Ketamine – The NMDAR Hypothesis and Beyond
Antidepressant effects are maintained long after ketamine’s complete metabolism and elimination (∼1 week vs. plasma half-life of 2–3 h 19 ), and therefore cannot be attributed simply to the drug’s ability to block NMDARs acutely. Instead the effects appear to be due to the activation of crucial downstream signalling cascades arising as a secondary consequence of NMDAR inhibition, resulting in long-lasting adaptations in key neural circuits.13,20–22 This suggestion reflects significant elaboration of the NMDAR hypothesis of ketamine action in recent years, as new evidence has emerged concerning events downstream of NMDAR inhibition.
Pre-clinical and clinical studies suggest that depression involves pathological glutamate excitotoxicity, synaptic dysfunction and neuronal atrophy in key brain areas, particularly the hippocampus (HPC) and prefrontal cortex (PFC).10,23–25 The NMDAR hypothesis of ketamine has given rise to two major, non-mutually exclusive, models to explain the antidepressant action of this compound, namely the ‘disinhibition’ and the ‘direct inhibition’ hypotheses.26,27 The first hypothesis (Figure 1(a)) proposes that low doses of ketamine selectively antagonise NMDARs on GABAergic inhibitory interneurons leading to disinhibition of excitatory pyramidal neurons (PNs), a burst of glutamate release and acute AMPAR activation in the PFC and HPC.
10
This, in turn, results in the activation of downstream signalling pathways, particularly those involving brain-derived neurotrophic factor (BDNF) release and activation of the mammalian target of rapamycin (mTOR).5,22,28,29 Under the second hypothesis (Figure 1(b)), direct antagonism of NMDARs on PNs by ketamine at rest is thought to block tonic NMDAR activation by ambient or spontaneously released glutamate (due to an incomplete Mg2+ block). This has been proposed to block glutamate excitotoxicity, reduce suppression of eukaryotic elongation factor 2 (eEF2)-mediated protein synthesis and recruit the downstream signalling pathways mentioned above (e.g. BDNF, mTOR).26,30 Consistent with a direct action on PNs, selective genetic deletion of the NMDAR subunit GluN2B from cortical PNs both mimics and occludes ketamine’s antidepressant effects in mice, as well as enhancing protein synthesis and AMPAR-mediated transmission in the PFC.
31
It has been proposed that low doses of ketamine selectively block GluN2B-containing NMDARs as they are thought to be (1) tonically activated by spontaneously released and/or ambient glutamate and (2) mainly extra-synaptic and potentially more accessible to exogenous antagonism, although this continues to be debated.26,32–34 At high doses, ketamine might gradually inhibit synaptic NMDARs, leading to dissociative effects and eventually anaesthesia.
26
This body of work has prompted the hypothesis that while ketamine is non-subunit specific, antagonism of GluN2B-containing NMDARs might be responsible for its antidepressant action.26,31,32 In partial support of this, the selective GluN2B antagonist, Ro25-698 possesses rapid antidepressant action in rodents, but the effects are reported to be less robust and/or shorter-lasting compared to ketamine.11–14,28,35
Schematic of three competing hypotheses of ketamine action in depression. (a) Disinhibition hypothesis of ketamine: (1) At low doses, ketamine preferentially inhibits NMDARs on GABAergic interneurons (INs), (2) reduced excitability of inhibitory INs causes disinhibition of glutamatergic pyramidal neurons (PNs), (3) leading to a surge of glutamate release, which binds to and activates postsynaptic AMPARs, (4) the AMPAR-mediated depolarisation activates voltage-gated calcium channels (VDCCs), (5) Ca2 + triggers the activity-dependent synaptic release of BDNF, which acts on its surface receptor TrkB, (6) to activate two major downstream signalling cascades (MEK-ERK and PI3K-Akt), which converge onto mTOR, (7) mTOR activation leads to disinhibition of synaptic protein translation (e.g. GluR1-2, PSD95) and (8) newly synthesised AMPARs and other synaptic components are inserted into the postsynaptic density. (b) Direct inhibition hypothesis of ketamine: (1) At rest, ketamine directly inhibits NMDARs on PNs, which can be tonically activated by ambient and spontaneously released glutamate (due to an incomplete Mg2 + block), (2) antagonism of tonically active (presumably extrasynaptic NR2B subunit-containing) NMDAR prevents glutamate excitotoxicity and blocks activation of the eukaryotic elongation factor 2 (eEF2) kinase, (3) which reduces suppression of eEF2 and (4) results in enhanced eEF2-mediated protein translation, particularly of BDNF, (5–8) (see steps 5–8 in (a)): briefly, downstream signalling pathways involving BDNF and mTOR are recruited, leading to enhanced protein synthesis and AMPAR upregulation. (c) Unifying model of ketamine action: The (1) disinhibition and (2) direct inhibition hypotheses of ketamine are non-mutually exclusive and likely complementary. A unifying theory postulates that ketamine’s mechanism of action as an antidepressant may depend on its ability to (3) recruit key intracellular signalling cascades (BDNF and mTOR) and (4) initiate a long-term potentiation (LTP)-like process involving acute AMPAR activation, and importantly, desuppression of synaptic protein synthesis and AMPAR subunit upregulation. (5) Ultimately, ketamine increases the ratio of AMPA to NMDA receptor throughput via directly blocking NMDARs and indirectly enhancing AMPAR function, leading to synaptogenesis and reversal of stress-induced synaptic dysfunction and neuronal atrophy in the PFC and HPC, brain areas highly implicated in MDD. Importantly, the ketamine metabolite (2R,6R)-HNK, which was recently reported to be necessary and sufficient for ketamine’s antidepressant effects, also seems to facilitate AMPAR-mediated transmission, while potentially indirectly inhibiting NMDAR function. However, the exact molecular target and mechanism of action of (2R,6R)-HNK remain unknown. Notably, the AMPAR antagonist NBQX abolishes ketamine and (2R,6R)-HNK’s sustained antidepressant effects, while AMPA potentiating agents (or AMPAkines) possess antidepressant action, further supporting the role of AMPARs. Figure adapted from Aleksandrova et al.
13
and Sanacora and Schatzberg
68
with permission.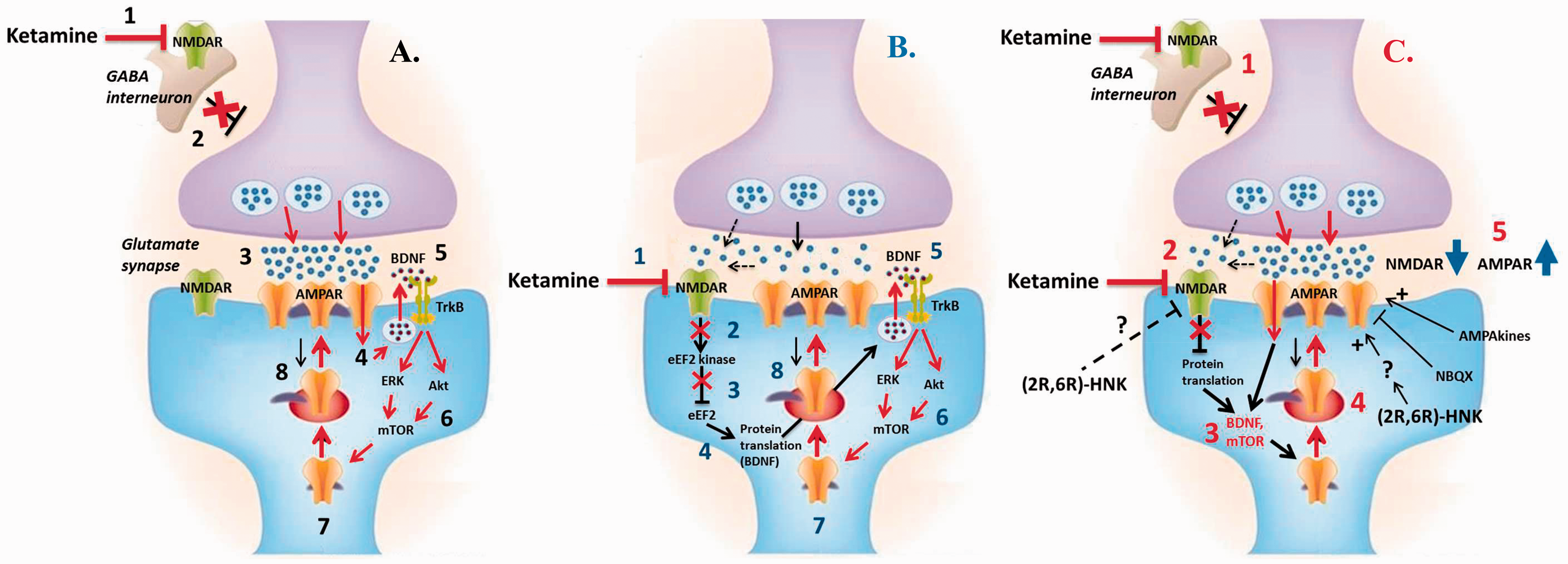
Regardless of the exact trigger, ketamine initiates a long-term potentiation (LTP)-like process leading to synaptic protein synthesis, AMPAR subunit upregulation and synaptogenesis in the HPC and PFC.28,36,37 Thus, it is thought to induce an NMDAR inhibition-dependent form of synaptic plasticity, reversing the stress-induced synaptic dysfunction and neuronal atrophy in brain areas implicated in MDD, effects believed to underlie the antidepressant response to ketamine.13,24,38 Together, these studies have significantly expanded on the simplified NMDAR inhibition hypothesis of ketamine action, supporting the crucial involvement of AMPARs and synaptogenic pathways in mediating ketamine’s antidepressant effects.10,13,28 A unifying theory (Figure 1(c)) postulates that ketamine’s mechanism of action in depression may depend on its ability to increase the ratio of AMPA to NMDA receptor throughput via directly blocking NMDARs and indirectly enhancing AMPAR function.13,20,22,28
The NMDAR inhibition hypothesis of ketamine in depression has inspired tremendous interest in alternative NMDAR antagonists, such as memantine, AZD6765 and CP-101,606, as novel, safer antidepressants. However, to date, ketamine is the only NMDAR antagonist to consistently demonstrate antidepressant efficacy in multiple trials.6,14,39 In addition, while ketamine is usually administered as a racemic mixture in the clinic, R-ketamine has more potent and sustained antidepressant properties in rodents compared to S-ketamine.39,40 This observation directly challenges the NMDAR hypothesis of ketamine antidepressant action, as R-ketamine is ∼4 times less potent at inhibiting NMDAR relative to S-ketamine, which is, in turn, is associated with more potent anaesthetic and psychotomimetic side effects.39–41 Findings like these appear to refute the NMDAR hypothesis of ketamine’s antidepressant effects.
(2S,6S;2R,6R)-Hydroxynorketamine
As noted above, the NMDAR hypothesis of ketamine action in depression was further challenged by the recent findings of Zanos et al. 14 focused on an active ketamine metabolite, (2S,6S;2 R,6 R)-HNK. Accordingly, a brief synopsis of ketamine’s metabolism may shed further light on this issue.
The Early Days of Ketamine Metabolomics
(R,S)-Ketamine is stereoselectively metabolised in the liver by microsomal enzymes into a wide range of metabolites, including (R,S)-norketamine (NK), two diasteromeric hydroxyketamine (HK) and six diastereomeric HNK metabolites, and (R,S)-dehydronorketamine (DHNK) (Figure 2).
42
An early study demonstrated that unlike ketamine and its primary metabolite (NK), the major secondary metabolites (2S,6S;2R,6R)-HNK and DHNK lack anaesthetic properties associated with NMDAR antagonism.
41
These findings led to the conclusion that ketamine’s secondary metabolites are inactive and do not contribute to ketamine’s clinical effects, an assumption that deterred subsequent characterisation of their pharmacological activity and would not be effectively challenged until recently.

Pharmacokinetic Considerations
Overall, the long-lasting antidepressant effects following a single ketamine infusion (∼1 week) are remarkable given the short half-life of ketamine (2–3 h).42,43 On the other hand, secondary ketamine metabolites including (2S,6S;2R,6R)-HNK have much longer (but also quite variable) plasma half-lives (16.5 and 7.7 h for (2R,6R)- and (2S,6S)-HNK, respectively) 44 and are still detectable at three days post-infusion 43 (no data available past this time point). Peak plasma (2S,6S;2R,6R)-HNK levels reached following administration of clinically relevant doses of ketamine in humans has been reported to be ∼0.1 μM, 43 whereas in rodents the levels in the brain are ∼1 μM following systemic injection of ketamine. 14 Intriguingly, clinical studies suggest that the conversion of (R,S)-Ket may be enantioselective, as significantly higher plasma concentrations of (2R,6R)-HNK relative to (2S,6S)-HNK are observed post-ketamine administration.42,44 One study in rats reports that sub-chronic ketamine dosing increased the clearance of (R,S)-Ket and (R,S)-NK, while producing significant increases in the plasma concentrations of (2S,6S;2R,6R)-HNK (∼4.5-fold), compared to a single administration. 45
Therefore, (2S,6S;2R,6R)-HNK is a major metabolite of ketamine with a considerably longer half-life, and thus, its potential contribution to ketamine’s clinical effects is a very appropriate line of research. However, it is clear that factors such as the species, route and frequency of ketamine administration, as well as individual variability (e.g. sex, genetics and disease state), 43 can alter the efficacy of ketamine metabolism, producing different metabolite patterns and potential differences in clinical effects. Route of administration is a particularly important factor, which almost always differs in human versus rodent studies of ketamine (IV vs. intraperitoneal, i.p.), and would greatly impact the pharmacokinetics of the drug, thereby limiting the ability to directly translate findings from rodents to humans. Ketamine is administered via the i.p. route in rodents, and therefore, undergoes extensive first-pass liver metabolism. As a consequence, whereas the bioavailability of the parent compound can be as low as 20%, plasma and brain concentrations of its metabolites would be relatively high.5,16 In contrast, ketamine is given clinically as an IV infusion, thereby greatly reducing first-pass metabolism, resulting in a different profile of ketamine and metabolite concentrations in the blood and brain.5,16 It is also important to note that unlike the parent compound, which rapidly accumulates in the rodent brain following systemic administration, (2S,6S;2R,6R)-HNK seems to exhibit the opposite pattern (ratio of max brain/plasma concentrations ∼0.5 vs. 1.75 for ketamine), which could additionally limit the contribution of metabolites to the central actions of ketamine. 46 In order to critically evaluate (2S,6S;2R,6R)-HNK’s contribution to ketamine’s antidepressant effects in the clinic, it is crucially important to determine the brain concentrations of (2S,6S)- and (2R,6R)- HNK following infusion of clinically relevant doses of ketamine in humans. Overall, although no significant relationship between plasma metabolite levels and ketamine response has been reported to date, secondary metabolites could contribute to ketamine’s sustained therapeutic activity, a possibility that warrants further evaluation.
Summary of Findings With (2R,6R)-HNK by Zanos et al.
Firstly, Zanos et al. replicate the finding that R-ketamine has more potent and sustained antidepressant effects compared to S-ketamine in several rodent tests of depression.14,39,40 Previous studies have also indicated that ketamine produces more robust antidepressant responses in female compared to male rodents.47,48 Similarly, Zanos et al. found that in females lower doses of ketamine are required to obtain significant reductions in depression-related behaviours in the forced swim test (FST), novelty-suppressed feeding test and learned helplessness (LH) test. 14 This observation prompted them to compare the pharmacokinetic profiles of ketamine and its metabolites in male and female mice. Importantly, although no sex differences in the plasma or brain levels of ketamine or NK are reported, brain concentrations of the most abundant metabolite, (2S,6S;2R,6R)-HNK, are three-fold higher in female mice, offering a possible explanation of ketamine’s enhanced potency in females. 14 This group also finds that a metabolically inert form of ketamine (6,6-dideuteroketamine), with the same apparent NMDAR binding and pharmacokinetic properties except for the capacity to be metabolised into (2S,6S;2R,6R)-HNK, lacks ketamine’s sustained (24 h) antidepressant actions in the FST and LH tests. 14 The seminal finding by Zanos et al. is that the (2R,6R)-HNK metabolite itself (10 mg/kg, i.p.) has robust rapid and sustained antidepressant action in these tests and also reverses social deficits induced by prior chronic social defeat stress. In all tasks, (2R,6R)-HNK is more potent than (2S,6S)-HNK, thereby mirroring the enantiomer-specific effects of the parent compound. 14 Overall, based on these novel findings, Zanos et al. claim that (2R,6R)-HNK is a key active metabolite of ketamine that is both necessary and sufficient for the antidepressant actions of its parent drug.
To reiterate, the fact that (2R,6R)-HNK’s antidepressant action appears to be NMDAR-independent poses the greatest challenge to the primacy of ketamine’s direct action on the NMDAR in accounting for its remarkable antidepressant effects. Specifically, unlike its parent compound (Ki for NMDAR = 465 nM and 1340 nM for (S)- and (R)-Ket, respectively), (2S,6S; 2R,6R)-HNK does not functionally inhibit NMDARs (Ki > 10 μM). 14 In agreement with this, a previous study reported that (2S,6S)- and (2R,6R)-HNK have low binding affinities for the NMDAR (Ki = 21 μM and > 100 μM, respectively). 41 Interestingly, Zanos et al. also show that bath application of 10 μM (2R,6R)-HNK causes a dramatic increase in AMPAR-mediated excitatory post-synaptic potentials (EPSPs, slope up by ∼600%) in hippocampal CA1 slices within an hour, which persists after washout. 14 Furthermore, in agreement with these electrophysiological data, systemic injection of both ketamine and (2R,6R)-HNK induces a selective increase in gamma band power in vivo as measured by quantitative electroencephalography, consistent with increased the activation of fast ionotrophic excitatory receptors (including AMPAR). 14 Similar to ketamine, a single dose of (2R,6R)-HNK also causes an upregulation of GluA1 and GluA2 AMPAR subunits, a decrease in eEF2 phosphorylation, as well as an increase in BDNF expression in hippocampal (but not PFC) synaptosomes 24 h after the treatment. 14 Similar to the findings with ketamine, the AMPAR antagonist NBQX administered either before (2R,6R)-HNK injection or at the time of behavioural testing, blocks the rapid and sustained antidepressant effects of the metabolite, further implicating AMPAR activation. 14 Accordingly, these results provide a compelling argument against the NMDAR hypothesis of ketamine and call for a focus on (2R,6R)-HNK and its effects on AMPARs as key to understanding ketamine’s antidepressant actions. However, the exact mechanisms underlying the actions of (2R,6R)-HNK on AMPAR-mediated transmission remain to be explored.
Importantly, in contrast to ketamine, (2R,6R)-HNK lacks sensory-dissociation, motor in-coordination and hyperlocomotion effects, as well as reinforcing properties in drug discrimination and self-administration protocols in rodents. 14 Therefore, (2R,6R)-HNK appears to lack psychotomimetic effects and abuse potential in preclinical tests and has a benign side effects profile compared to ketamine, presumably because of the lack of activity at the NMDAR.
Critique and Controversy
These breakthrough findings created great interest, prompting a re-evaluation of the NMDAR hypothesis of ketamine, and highlighting (2R,6R)-HNK as a promising new, safer, rapid-acting antidepressant. As may be expected, however, this discovery has come under close scrutiny as exemplified by a recent commentary by Collingridge et al., 18 cautioning against rejection of the NMDAR hypothesis of ketamine action. The finding that the NMDAR antagonist MK-801, which binds to the same receptor site as ketamine, lacks ketamine’s sustained antidepressant effects, 14 was taken by Zanos et al. as further evidence against the NMDAR hypothesis of ketamine. However, as Collingridge et al. rightfully point out, there are key differences in how the two drugs interact with the NMDAR site, which could account for the observed differences in clinical activity. 18 Both MK-801 and ketamine are open-channel blockers that only bind to the open state of the receptor, which appears to be a crucial feature for ketamine’s mechanism of action, as it allows for a preferential block of excessive activation over normal synaptic activity. 49 However, unlike ketamine, MK-801 has a high affinity, is almost completely trapped inside the receptor once pore closes and has a slow dissociation rate (time constant > 3 min), making it highly neurotoxic.18,50,51 On the other hand, memantine, a low-affinity, partial-trapping (50%–70%) NMDAR antagonist with a fast off-rate (∼3 s), lacks significant side effects, yet it also lacks antidepressant action due to relatively rapid kinetics.18,50,51 Importantly, ketamine binds to the NMDAR with a low affinity and high but not complete trapping (86%), and is uniquely associated with the induction of NMDAR inhibition-dependent synaptic plasticity, which is thought to underlie its unique antidepressant effects.14,32 The differential clinical action of NMDAR blockers can be attributed to differences in the nature of NMDAR block, and therefore, the absence of antidepressant properties of one member of this class of drugs (i.e. MK-801) should not be taken as evidence against the NMDAR hypothesis of ketamine. On the other hand, Collingridge et al. also emphasise that other NMDAR blockers have rapid and/or sustained antidepressant effects, including CP-101,606 (selective negative allosteric modulator of GluN2B subunit-containing NMDARs), Mg2+ and glycine-cite NMDAR antagonists. 18 A final question concerns whether the effects of (2R,6R)-HNK on AMPAR-mediated transmission in vitro, as well as on tests of depression-related behaviours in vivo, 11 would be observed with clinically relevant doses of ketamine. 18 Indeed, the (2R,6R)-HNK concentration used by Zanos et al. in their electrophysiology experiment (10 μM), as well as the maximum concentration reached following a systemic injection of (2R,6R)-HNK (10 mg/kg, i.p., 10.69 μM), 14 are ∼10–100 times higher than peak plasma metabolite levels obtained following administration of clinically relevant doses of ketamine in mice (∼1 μM) 14 and in humans (∼0.1 μM). 43
In their rebuttal, 52 Zanos et al. again point out that alternative NMDAR antagonists have generally failed to recapitulate ketamine’s potent, rapid and sustained antidepressant effects, and to date, ketamine is the only NMDAR antagonist to consistently demonstrate antidepressant efficacy in multiple trials.6,11–14,28,39 Furthermore, they emphasise that (2S,6S;2R,6R)-HNK levels in the brain of humans undergoing ketamine infusions are unknown, and because of this, as well as significant pharmacokinetic and pharmacodynamic differences between humans and rodents (e.g. route of administration), direct comparisons with clinically relevant concentrations are not possible. In addition, Zanos et al. postulate that concentrations lower than 10 μM may well produce the same effect on AMPAR-mediated transmission, although to a lesser extent. However, it is important to note that this experiment involved bath application of (2R,6R)-HNK onto hippocampal slices, which has crucial limitations (e.g. lack of intact brain circuitry, difficulty in mimicking physiological conditions, no normal distribution, metabolism, elimination of drug). Thus, it is crucial that future studies evaluate the effects of (2R,6R)-HNK and ketamine, on basal synaptic transmission, as well as synaptic plasticity (LTP and long-term depression, LTD), in vivo and in different brain areas implicated in depression. Such experiments will also address an apparent lack of studies on the effects of ketamine on synaptic plasticity processes, effects which could provide a far-reaching mechanism to explain how ketamine’s molecular and cellular effects translate into region-specific changes in structural plasticity and neural circuit functioning.
Hashimoto also published a recent commentary 15 discussing the implications and limitations of the Zanos et al. study. Importantly, he emphasises that in their study the ‘sustained’ antidepressant effects of ketamine and (2R,6R)-HNK are assessed at 24 h after injection; however, authors did not test for a longer-lasting antidepressant effect (seven days), as is commonly done in studies of ketamine. 15 Another inconsistency in the Zanos et al. paper is that the molecular effects of ketamine and (2R,6R)-HNK (e.g. BDNF and GluR1,2 upregulation) were only detected in the HPC but not in the PFC, 14 which is in contrast with a body of literature implicating the PFC as key mediator of ketamine’s antidepressant effects.13,15,30,52–54 Moreover, Hashimoto points out that the higher potency of ketamine in females could be attributed to factors other than the higher brain levels of (2S,6S;2R,6R)-HNK, including the contribution of gonadal hormones, which have been shown to potentiate ketamine’s antidepressant effects.15,19,47,48 Importantly, the enhanced activity of ketamine is abolished in ovariectomised females and further restored by estrogen and progesterone replacement. 48
It is also important to note that despite the fact that mTOR has been repeatedly implicated in ketamine’s antidepressant mechanism,13,29,36,55,56 Zanos et al. did not detect any changes in mTOR phosphorylation following administration of either (2R,6R)-HNK or ketamine. 14 In fact, an earlier study by the same group57 found that (R,S)-ketamine (40 mg/kg, i.p.) and (2S,6S)-HNK (20 mg/kg, IV) induced a significant time-dependent increase in mTOR phosphorylation in the PFC. 57 However, differences in the enantiomer, brain area, dose and route of administration used prevent direct comparison with the results of Zanos et al. and leave uncertainty as to (2S,6S;2R,6R)-HNK’s effects on mTOR signalling, a pathway known to contribute to ketamine’s action.
Lastly, Zanos et al. claim that ‘published human data reveal a positive correlation between the antidepressant responses of ketamine and plasma (2S,6S;2R,6R)-HNK metabolite levels.’ 14 It is important to note that this statement is somewhat inaccurate, as the cited 2012 study from the same group 43 demonstrated that while ketamine responders tended to have higher plasma (2S,6S;2R,6R)-HNK metabolite levels than non-responders, this correlation was not significant (p = 0.88). 43
The claim that (2R,6R)-HNK does not bind NMDARs was disputed by Suzuki et al. in a recent brief communication. 16 Specifically, while 10 µM (2R,6R)-HNK did not alter NMDA-mEPSCs in hippocampal slices, 50 µM produced a significant inhibition of NMDAR currents at rest (∼40% vs. almost 100% with 50 µM ketamine), and only this higher dose was found to induce a decrease in eEF2 kinase phosphorylation. 16 Based on these results, they conclude that similar to its parent compound, (2R,6R)-HNK is an open-channel NMDAR antagonist, which converges onto the same intracellular signalling pathways previously implicated in ketamine’s antidepressant action, namely eEF2k inhibition, increased BDNF and AMPAR upregulation in the HPC.14,16 Accordingly, authors propose that both ketamine and (2R,6R)-HNK involve a form of NMDAR-induced AMPAR-dependent synaptic potentiation. 16
In response, Zanos et al. offered a further rebuttal 52 insisting that, in fact, these latest findings do not contradict but rather complement their own results, because no significant NMDAR binding was observed for (2R,6R)-HNK at doses up to 10 μM in both studies, indicating no clinically relevant activity at this receptor. 52 This is in agreement with the available data, which indicate that (2R,6R)-HNK concentrations in the brain likely translate to the magnitude of ∼0.1–1 μM, although as mentioned, considerable debate exists over this point due to the lack of human data and difficulties in translating data from rodents to humans.14,17,32,34,43 Overall, despite this controversy, data from different laboratories seem to agree that (2R,6R)-HNK does not possess NMDAR inhibitory properties at therapeutically relevant concentrations.
Adding to the controversy surrounding (2R,6R)-HNK, Yang et al. failed to replicate the antidepressant effects of the ketamine metabolite in two rodent models of depression, the lipopolysaccharide injection and the chronic social defeat stress models. 17 Although a single dose of (R)- and (S)-ketamine (10 mg/kg, i.p.) had rapid and sustained antidepressant effects in the FST, tail suspension test and the sucrose preference test, an equivalent dose of (2R,6R)-HNK failed to alleviate both the inflammation- and chronic stress-induced depression-like phenotypes. 17 No apparent reason for the discrepancy in findings between the two groups can be identified.
In order to settle the controversy currently surrounding the behavioural effects of this metabolite in rodents, it is crucial that (2R,6R)-HNK be systematically evaluated in animal models of depression with high predictive, face and construct validity, particularly the chronic mild stress paradigm and the Wistar-Kyoto rat (an endogenous model of depression).58–60 Another general limitation relates to the prevalence of MDD in women, which is approximately two times higher than in men; yet despite this important sex difference, preclinical research investigating the mechanisms underlying ketamine’s, and more recently (2R,6R)-HNK’s, antidepressant effects have been conducted almost exclusively in male rodents.13,23 It is crucial that future animal studies of antidepressant compounds, particularly ketamine and (2R,6R)-HNK, which exhibit dramatic sex differences in efficacy,14,47,48 should include both sexes. Importantly, in this regard, the Wistar-Kyoto strain of rats offers a marked advantage, as females have a more pronounced depressive-like phenotype than males, similar to MDD.61,62
In light of the breakthrough, but controversial, findings of Zanos et al., questions remain as to whether (2S,6S;2R,6R)-HNK mediates, or at least contributes to, ketamine’s antidepressant effects, and its implications for the NMDAR hypothesis of ketamine. Further research is clearly required to replicate its antidepressant effects, to determine its molecular targets and exact mechanism of action and to evaluate its potential as a novel antidepressant in the clinic.
(2R,6R)-HNK’s Potential Mechanism of Action
As (2S,6S;2R,6R)-HNK does not apparently inhibit NMDARs at therapeutically relevant concentrations, the molecular target(s) responsible for its behavioural and synaptic effects are still unclear. Importantly, one previous study reported that (2R,6R)- and (2S;2R)-HNK inhibit α7-nicotinic acetylcholine receptor (nAchR) function at concentrations <1 μM using patch-clamp techniques. 41 At the synapse, α7-nAChRs induce presynaptic glutamate release, as well as increase ambient extrasynaptic glutamate by activating glutamate release from astrocytes. 63 Alpha7-nAChR inhibition prevents Ca2+ influx, which in turn may reduce glutamate release and excitotoxocity, as well as alter the activation of downstream signalling pathways. 64 It is currently unclear whether the (2R,6R)-HNK-induced synaptic and molecular effects14,16,57,64 are related to its inhibitory effects on neuronal and/or astrocytic α7-nAChRs. Here, it is important to note that clinical studies have supported the utility of open-channel non-competitive α7-nAchR inhibitors for the treatment of depression. 41
Another previous study reported significant effects of ketamine metabolites on D-serine concentrations in vitro, highlighting another possible mechanism of action for (2R,6R)-HNK. 64 D-serine is an endogenous NMDAR co-agonist, which can be elevated under pathological conditions and has been linked to NMDAR-mediated neurotoxicity and neurodegeneration.64,65 Cell culture incubation with (2S,6S)- and (2R,6R)-HNK attenuated both extracellular and intracellular D-serine concentrations in a dose-dependent manner (max inhibition ∼50%; IC50 = 0.18 nM and 0.68 nM, respectively).64,65 The reduction in D-serine levels is attributed to α7-nAchRs inhibition by (2S,6S;2R,6R)-HNK, which is thought to attenuate the Ca2+-dependent synthesis of D-serine.57,64,65 Although (2R,6R)-HNK does not directly inhibit NMDARs at clinically relevant concentrations, it is possible that its mechanism of action involves indirect NMDAR inhibition elicited by a reduction in D-serine co-agonist levels, a theory that awaits validation in vivo. 64 Importantly, this hypothesis is supported by clinical reports of a link between antidepressant response to ketamine and plasma D-serine concentrations, showing that responders had significantly lower pre-treatment D-serine levels and ketamine further resulted in significant reductions over the next 24 h, attributed to the action of ketamine metabolites. 66 It is possible that (2R,6R)-HNK’s facilitatory effects on AMPAR-mediated transmission, intracellular signalling pathways and protein synthesis are due to this indirect NMDAR inhibition, representing a point of convergence with ketamine, or due to some other molecular targets of HNK yet unknown. Despite the significant gap in current understanding of (2R,6R)-HNK’s mechanism of action, available data support a hypothesis that like ketamine, (2R,6R)-HNK might be able to increase the AMPA to NMDA receptor throughput (Figure 1(c)), although future studies are required to define (2R,6R)-HNK’s molecular target(s) and exact mechanism of action, and importantly, determine to what extent they contribute to ketamine’s antidepressant action.
Conclusions – (2R,6R)-HNK, Ketamine and the NMDA Hypothesis
It is clear that ketamine’s clinical efficacy in depression cannot be solely attributed to its acute NMDAR antagonism. The prevailing hypothesis of ketamine action now implicates NMDAR inhibition-dependent synaptic plasticity that involves increased AMPAR function and activation of synaptogenic pathways in mediating ketamine’s antidepressant effects. However, the recent seminal study by Zanos et al. directly challenges the NMDAR hypothesis of ketamine with the claim that an active ketamine metabolite without NMDAR binding properties or key side effects of its parent compound is both necessary and sufficient for ketamine’s antidepressant effects in rodents. 14 However, following these encouraging initial findings, one group failed to replicate the antidepressant effects of (2R,6R)-HNK, 17 while others question the metabolite’s contribution to ketamine’s therapeutic effects or argue against rejecting the NMDAR hypothesis of ketamine action.15–18
Although the hypothesis that NMDAR blockade is solely responsible for ketamine’s antidepressant effects has long been challenged; rejecting the role of NMDARs altogether is premature. The claim that ketamine’s clinical efficacy is exclusively due to (2R,6R)-HNK continues to receive considerable resistance in the field and should be taken with caution.15–18 The major concern regarding the contribution of (2R,6R)-HNK to ketamine’s clinical effects is whether clinically relevant doses of ketamine could achieve the brain levels of (2R,6R)-HNK(∼10 μM) required for its reported antidepressant, molecular and synaptic effects.17,18,27 Another general limitation prompting the field to question the relevance of preclinical findings with (2R,6R)-HNK to clinical situations is the difficulty in translating findings from rodents to humans given the different routes of administration involved. In rodents, ketamine is administered via an i.p. injection, and that will undoubtedly exaggerate the contribution of metabolites to its antidepressant effects due to extensive first-pass metabolism, which would not occur following an IV infusion in humans. 16 In addition, strong evidence against a role for metabolites in ketamine’s antidepressant effects comes from the finding that a single, bilateral microinfusion of (R)-ketamine directly into the mPFC or HPC mimicked the effects of systemic administration, indicating clearly that ketamine itself can exert antidepressant effects. 67 Therefore, despite the finding that 6,6-dideuteroketamine (not metabolised into (2S,6S;2R,6R)-HNK and with all other pharmacokinetic and receptor properties apparently unaltered) lacks ketamine’s sustained antidepressant effects, 14 a growing consensus questions whether (2R,6R)-HNK can be solely responsible for ketamine’s clinical effects.15–18,67
Findings with (2R,6R)-HNK do, however, represent important progress in the field and are the latest piece of the ketamine puzzle. The possibility that a key metabolite previously deemed inactive could mediate, or at least contribute to, the antidepressant effects of ketamine has revised the way we think about and probe ketamine’s mechanism of action in depression. It is clear that (2R,6R)-HNK represents a major plasma metabolite with a considerably longer half-life, which could very well contribute to the remarkably long-lasting antidepressant effects following a single ketamine infusion.14,42,43 Although the molecular target(s) of (2R,6R)-HNK have not been defined, studies have implicated increased AMPAR-mediated synaptic transmission, BDNF and protein synthesis,14,57 which seem to represent points of convergence with ketamine.10,22,28 It is also important to consider other, unique effects of (2S,6S;2R,6R)-HNK such as α7-nAchR antagonism 41 and attenuation of D-serine levels, which could possibly lead to an indirect inhibition of NMDAR function. 64 Therefore, (2R,6R)-HNK could contribute to ketamine’s unique ability to increase the AMPA to NMDA receptor throughput by indirectly inhibiting NMDARs and enhancing AMPAR function (Figure 1(c)). In turn, these effects converge onto downstream synaptogenic signalling pathways, thereby restoring synaptic strength and connectivity in key brain areas (HPC and PFC). 13 Thus, the possible contribution of metabolites to ketamine’s clinical effects remains a distinct possibility and should be further examined in the future.
In general, a critical evaluation of (2S,6S;2R,6R)-HNK’s contribution to ketamine’s clinical activity depends on a better understanding of its pharmacokinetics and pharmacodynamics, which determine (1) how much (2S,6S)- and (2R,6R)-HNK is present in the brain (brain Cmax) following infusion of clinically relevant doses of ketamine in humans and (2) what are the molecular targets and the corresponding affinity of (2S,6S)- and (2R,6R)-HNK (IC50, Ki values), indicating whether its effects would be relevant at therapeutic doses of ketamine. Currently, there are no conclusive data to provide answers to these questions; however, emerging data will provide more insight into (2S,6S;2R,6R)-HNK’s contribution to ketamine’s effects in depression.
In addition to the findings with (2R,6R)-HNK, there seem to be two major challenges to the NMDAR hypothesis of ketamine in depression. Firstly, Zanos et al. argue that the general failure of alternative NMDAR antagonists as antidepressants6,11–14,28,39 should be taken as strong support against the NMDAR hypothesis of ketamine. However, as discussed, ketamine possesses several crucial characteristics (i.e. relatively low affinity, high but not complete trapping, non-subunit selective, non-competitive and open-channel nature of block), which make it unique within the broad class of NMDAR inhibitors. Thus, the unique nature of ketamine’s interaction with its target result in an optimal level of NMDAR inhibition (likely <50% NMDAR block under physiological conditions) 27 compared to other antagonists and likely explains why NMDAR antagonism is not universally antidepressant. It is possible that the same argument can be applied to the apparently contradictory enantiomer potency in depression, which represents the other major challenge to the NMDAR hypothesis of ketamine. As mentioned, R-ketamine has more potent and sustained antidepressant effects in rodents, even though it is ∼4 times less potent at inhibiting NMDAR relative to S-ketamine.14,39–41 Here, it is crucial to note that all clinical trials of ketamine to date have used one dose (0.5 mg/kg), and thus, the optimal dose of ketamine for TRD is still unknown. Similarly, animal studies have almost exclusively utilized a single ketamine dose (10 mg/kg), and the limited dose–response data available are inconclusive.14,28 Although purely speculative at this point, it is possible that at the doses tested, R-ketamine achieves a more favourable level of NMDAR inhibition compared to S-ketamine, which due to its high affinity might disrupt normal NMDAR-mediated synaptic plasticity, leading to a higher risk of side effects, but also diminished therapeutic efficacy. Clearly, rejecting the NMDAR hypothesis of ketamine action altogether is premature, but further investigation is clearly warranted in order to clarify the contribution of NMDAR blockade to ketamine’s unique antidepressant activity.
Important questions remain as to what extent (2R,6R)-HNK contributes to ketamine’s therapeutic actions, and given the conflicting behavioural data, whether it represents a new promising candidate antidepressant compound; questions which warrant further preclinical and clinical investigation. In addition, future studies should be aimed at elucidating its molecular targets and exact mechanism of action, in the search for novel antidepressant targets. Despite conflicting results in rodents, a direct comparison between ketamine and (2R,6R)-HNK in depressed patients is of great interest, and a validation for the clinical use of (2R,6R)-HNK in humans for mood and anxiety disorders is underway at the US National Institute of Mental Health.17,52 Studies like these are particularly important at a time when the NMDAR hypothesis of ketamine’s antidepressant actions is hotly debated. Undoubtedly, a major contribution of the work to date on the unique antidepressant properties of ketamine lies in its heuristic influence. Great strides are being made towards a fuller understanding of the role of ionotrophic glutamate receptors in the etiology and treatment of depression. It is also quite apparent that uncovering the molecular mechanisms underlying the therapeutic effects of ketamine and possibly (2R,6R)-HNK, will aid the development of novel and more efficacious antidepressant agents so urgently needed to address a major public health concern, and could hold potential for the treatment of other stress-related psychopathologies, including bipolar disorder, post-traumatic stress disorder and suicidality.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Y. T. W. and A. G. P. declare a patent related to glutamate receptor function (A Peptide that Specifically Blocks Regulated AMPA Receptor Endocytosis and Hippocampal CA1 Long-term Depression; European 04789721.0, and United States 13/066,700). A. G. P. also declares a pending patent for the use of d-Govadine in the treatment of cognitive deficits.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funded by grants from the Canadian Institutes of Health Research to A. G. P. and Y. T. W. L. R. A. is supported by a graduate fellowship from the University of British Columbia.