
Abstract

Commentary on: Hare BD, Shinohara R, Liu RJ, Pothula S, DiLeone RJ and Duman RS. Optogenetic stimulation of medial prefrontal cortex Drd1 neurons produces rapid and long-lasting antidepressant effects. Nature communications. 2019; 10: 223.
The discovery of ketamine’s antidepressant effects has generated great excitement due to the rapid and sustained time course (within hours, lasting up to seven days), efficacy in treatment resistant individuals, 1 as well as a pharmacological profile that is distinct from traditional antidepressants. 2 Efforts have been ongoing to identify the molecular, cellular, and circuit mechanisms underlying the actions of ketamine. Regarding circuitry, dysfunction of the medial prefrontal cortex (mPFC) subregions, including the subgenual and anterior cingulate, have been implicated in depression, and preclinical models have been utilized to study the role of mPFC in depression-like behaviors.
Ketamine is an NMDA antagonist yet it produces a burst of glutamate in the mPFC following administration. Preclinical studies have demonstrated that mPFC activity following ketamine administration is necessary for ketamine’s rapid antidepressant effects, and that activation of principal neurons in the mPFC can produce persistent antidepressant responses. 3 These findings indicate that neurons and circuits within the mPFC may represent key targets in depression. Through interaction with downstream targets, the mPFC plays a role in the generation of numerous behaviors as well as the response to stress. This diversity of action is mediated by principal neurons projecting to numerous downstream targets. Notably, mPFC principal neurons are heterogeneous and have been characterized based on their projection targets, dendritic morphology, and response to neuromodulators. 4 Although the burst of glutamate following ketamine administration may excite many types of mPFC pyramidal neurons indiscriminately, we hypothesized that the antidepressant response may be carried by a discriminable population.
To probe this hypothesis, we utilized Cre-dependent expression of neuronal effectors in mouse lines carrying the Cre transgene in neurons expressing the D1 dopamine receptor (Drd1-Cre) or the D2 dopamine receptor (Drd2-Cre).
5
This approach had previously been demonstrated to allow cell-type-specific access to distinct populations of mPFC pyramidal cells.
6
We observed that utilizing a channelrhodopsin vector to allow optogenetic stimulation of Drd1 containing cells produced a rapid (observable 24 h after stimulation) and sustained (observable seven days after stimulation) antidepressant response (Figure 1). These effects are similar to ketamine and indicate that the circuit function responsible for depression-like behavior was rapidly and persistently altered by the prior period of stimulation. In contrast, stimulation of the Drd2 cell type did not produce an antidepressant response. Additional experiments produced convergent evidence for the importance of the Drd1 cell type to the antidepressant response. Chemogenetic inhibition of mPFC Drd1 expressing cells blocked the antidepressant response to ketamine, and D1 antagonist administration into the mPFC when ketamine was administered also eliminated the ketamine response. Notably, using a similar approach, we did not observe the Drd2 cell type to be necessary for ketamine-associated antidepressant effects. Finally, we established that mPFC Drd1 projections to the basolateral amygdala (BLA) were necessary and sufficient for generating the rapid antidepressant responses to stimulation of these neurons in mPFC (Figure 1).
Experimental design and observed outcome following optogenetic stimulation of defined pyramidal cell subtypes in the mPFC. Photostimulation of Drd1 containing neurons in the mPFC produced rapid antidepressant effects (24 h) that persisted for up to seven days. Photostimulation of mPFC Drd1 terminals in the BLA produced similar effects identifying this area as likely site of stimulation associated synaptic plasticity. BLA: basolateral amygdala; mPFC: medial prefrontal cortex.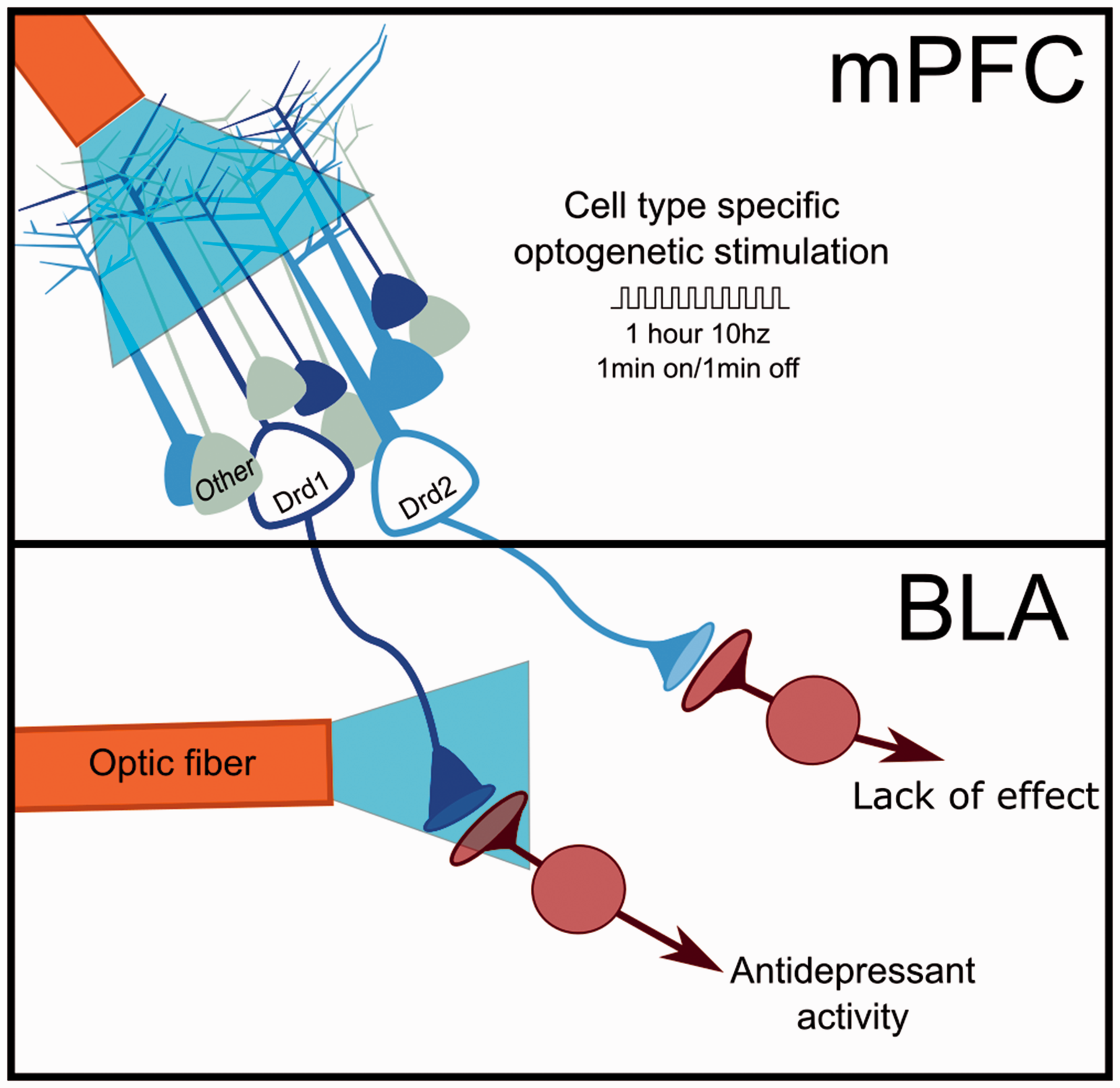
Depression is increasingly understood to be a disorder that is characterized by dysfunction of neural circuits and the synapses that directly produce intra- and inter-region communication. Numerous technologies currently exist that modulate brain activity to treat depression. These may be noninvasive such as transcranial magnetic stimulation or invasive in the case of deep brain stimulation. None of these technologies is capable of the type of defined neuronal control allowed by cell-type specific viral mediated expression vectors. However, we are not currently, nor are we likely in the near future, able to modulate human brain circuitry in a targeted fashion. What then is the utility of the type of approach we employed? Our hope is that continued study using these techniques will identify specific synaptic targets, and their modulators, that reverse antidepressant phenotypes in a way that is not achievable with the indiscriminate circuit modulatory approaches available for human use. Recent preclinical work in the field of addiction has demonstrated enhanced deep brain stimulation effects with coadministration of pharmacological agents and produced a framework for causally linking plasticity produced by circuit manipulations to changes in behavior.7,8 Key to this framework is identifying the plasticity associated with the disorder of interest and rationally designing therapeutically effective reversal protocols.
Our efforts point to a pair of synaptic targets that may offer antidepressant potential. The first is the D1 dopamine receptor within the mPFC. Prior research has demonstrated that systemic ketamine administration produces an increase in glutamate and dopamine within the mPFC. 9 Glutamatergic activity following ketamine has been associated with AMPA receptor activation, brain-derived neurotrophic factor release, and increased mTORC1 signaling within 30 to 60 min (for review, see Hare et al. 10 ). Increases in synaptic proteins (e.g., GluA1 and PSD95), spine density, and excitatory post synaptic currents, along with sustained antidepressant behavioral responses (up to seven days) 2 follow the immediate effects of ketamine. Our results demonstrate that mPFC-targeted D1 antagonist infusion blocks the antidepressant response to ketamine, and that mPFC D1 agonist infusion produces an antidepressant response. These findings are consistent with prior research demonstrating that D1 receptor activation concurrent with glutamatergic signaling through NMDA and AMPA receptors facilitates synaptic strengthening, similar to that observed after ketamine administration.11–13 Adjunctive antidepressant treatment with dopaminergic agents (e.g., aripiprazole, brexpiprazole) is approved in certain conditions. However, to our knowledge, there have been no efforts to determine whether these compounds would modulate ketamine’s antidepressant efficacy. Additionally, it is clear that rapid acting antidepressants rapidly reverse synapse loss associated with chronic stress models of depression, 14 and that mPFC photostimulation can produce synaptogenesis. 3 However, it is unclear whether mPFC Drd1 cell activation as we performed is sufficient to produce mPFC synapse formation or reverse the effects of chronic stress in animal models of depression.
The second synaptic target implicated by our work is the BLA, a key target of mPFC Drd1 neurons. We observed that photostimulation of mPFC Drd1 terminals in the BLA produced antidepressant-like effects well after the photostimulation period. It is currently unclear how mPFC-BLA synapses are altered by the stimulation protocol we employed, and whether the alteration would represent a reversal of pathological synaptic dysfunction that is produced in models of depression. To develop the described project into a translatable therapeutic approach, future studies will need to identify the specific BLA synaptic partners of mPFC Drd1 neurons, seek to understand how communication at these synapses is regulated in animal models of depression, and determine how the synaptic changes occurring following photostimulation impact pathological behavior.
These exciting, tractable, questions provide opportunities to develop novel insight into the circuit mechanisms underlying depression. Additionally, depression is not the only condition associated with circuit pathology. A better understanding of brain stimulation protocols and the plasticity they produce is likely to positively impact numerous pathological conditions such as addiction, schizophrenia, and obsessive compulsive disorder. These psychiatric conditions have enormous personal and societal impact, and continued development of treatments with circuit level mechanisms offers hope for restoring function in afflicted individuals and reducing disease burden.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: RSD has consulted and/or received research support from Naurex, Lilly, Forest, Johnson & Johnson, Taisho, and Sunovion. BDH declares no competing interests.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the NIMH (R01 MH105910-04 and RO1 MH093897-06A1 to R. S. D.) and a P&S Fund donation to the Brain and Behavior Research Foundation (NARSAD to B. D. H.).