
Abstract
Background
Patients with von Hippel-Lindau syndrome (VHL) have an increased risk of developing multiple neoplasms. Recently, multiple pharmacologic interventions have been assessed for the treatment of these VHL-associated neoplasms.
Objectives
To identify current clinical trials evaluating pharmacological interventions in VHL-associated neoplasms, with an emphasis in renal cell carcinoma (RCC).
Methods
We conducted a systematic review of the literature utilising MEDLINE/PubMed and EMBASE databases. We searched for Clinical Trials in VHL and RCC to understand the current landscape of therapeutic interventions in this population.
Results
We identified five single-arms clinical trials assessing systemic interventions in patients with VHL and RCC. These therapeutic interventions consisted of three tyrosine kinase inhibitors (TKIs) – semaxinib, sunitinib, and pazopanib - and one hypoxia-inducible factor (HIF) inhibitor -belzutifan. Belzutifan therapy was associated with an overall response rate (ORR) of 49% in patients with VHL and RCC. Only 3% of patients experienced disease progression while on belzutifan, which resulted in an impressive 97% clinical benefit rate. Pazopanib was also associated with an ORR of 64%; no patients experienced disease progression while on therapy. Lastly, two studies investigated the role of sunitinib in patients with VHL and RCC. In these studies, sunitinib was associated with an ORR ranging from 33% to 66%.
Conclusions
Anti-angiogenic interventions such as TKI and HIF inhibitors have been shown to be effective in decreasing the rate of progression of VHL-associated neoplasms. Although only a few trials have evaluated different pharmaceutical interventions in VHL-associated neoplasms, understanding the molecular basis of this pathology has opened the opportunity for novel therapeutic approaches to improve outcomes in this population.
Background
Von Hippel-Lindau (VHL) is an autosomal dominant syndrome characterised by the development of multiple benign and malignant tumors within different organs. The most commonly affected sites are the central nervous system (CNS), with the development of craniospinal and retinal hemangioblastomas seen in approximately 60% of patients, and the kidneys, with approximately 25%-60% of patients developing renal cell carcinomas (RCC). 1 Other common manifestations of these conditions are the presence of pheochromocytomas and pancreatic neuroendocrine tumors (PNET). 2 Historically, RCC has been the primary source of mortality in patients with VHL syndrome. 3 However, with the implementation of guided surveillance strategies, the incidence of metastatic RCC has decreased, leading to an overall decrease in mortality in patients with VHL syndrome.4–6 Yet significant morbidity can develop due to renal insufficiency as a consequence of frequent interventions to the kidneys, from partial or radical nephrectomies, focal radiation therapy, or cryotherapy.
Molecularly, VHL results from a germline mutation in the VHL gene, which functions as a tumor suppressor gene. This gene plays a crucial role in the oxygen-sensing pathway by regulating the proteasomal degradation of the α subunits of the hypoxia-inducible factor (HIF), which plays an essential role in angiogenesis in response to hypoxia. 7 Increased level of HIF acts as a transcription factor with subsequent increase in the production of multiple pro-tumorigenic molecules such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), multidrug resistance pup (MDR-1) and erythropoietin (EPO). 8
In patients with mutated VHL, there is a loss of function of the VHL protein, which mimics a hypoxic environment. In this setting, HIF- α accumulates and ultimately translocates to the cellular nucleus, where it increases the production of EPO, VEGF, PDGF, and TGF- α, leading to upregulation of angiogenesis and cellular proliferation.9,10 In VHL syndrome, the germline mutation of VHL is followed by the inactivation of the wild-type allele by epigenetic silencing or somatic mutation, leading to the loss of function of this protein and the cascade of events previously outlined. 11 A similar molecular signature has been observed in patients with non-familial clear cell RCC, where a somatic mutation of VHL is present in 46–82% of sporadic cases and is the hallmark of tumorigenesis. 8 The understanding of this biological process has led to the investigation of multiple pharmaceutical interventions with the goal of delaying tumor growth in patients with VHL (Figure 1). We performed a systematic review of the literature to define the current systemic pharmacologic therapeutic interventions in VHL, with an emphasis on VHL-associated RCC.

Role of VHL and target of therapeutic options in renal cell carcinoma.
Materials and methods
This study did not involve direct patient information, and evaluable data were obtained from publicly available sources utilising de-identified patient information. As such, this study did not require approval from the institutional review board.
We performed a systematic literature search utilising MEDLINE/PubMed and EMBASE databases. The search search strategy was developed for MEDLINE/PubMed and EMBASE using keywords ‘Renal Cell Carcinoma’, and ‘Von Hippel-Lindau’. Search strategy can be seen in Table 1. One independent reviewer (GGF) performed the search and evaluated all results. After the initial search was conducted, the data were reviewed by the senior reviewer (BLM) for accuracy. This systematic review was performed according to the recommendation of the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA). We limited our search to clinical trials published between January 2003 and December 2023 (Figure 2). For the purpose of this systematic review, non-clinical trial publications such as review articles, editorials, case reports, case series, and cohort studies were not included. Only systemic pharmacologic interventions were included. Given the paucity of randomised clinical trials (RCT) in VHL, non-randomised clinical trials were included. The data extracted from this search were tabulated using Microsoft Excel.

Flow diagram depicting our search strategy.
Search strategy.

Results
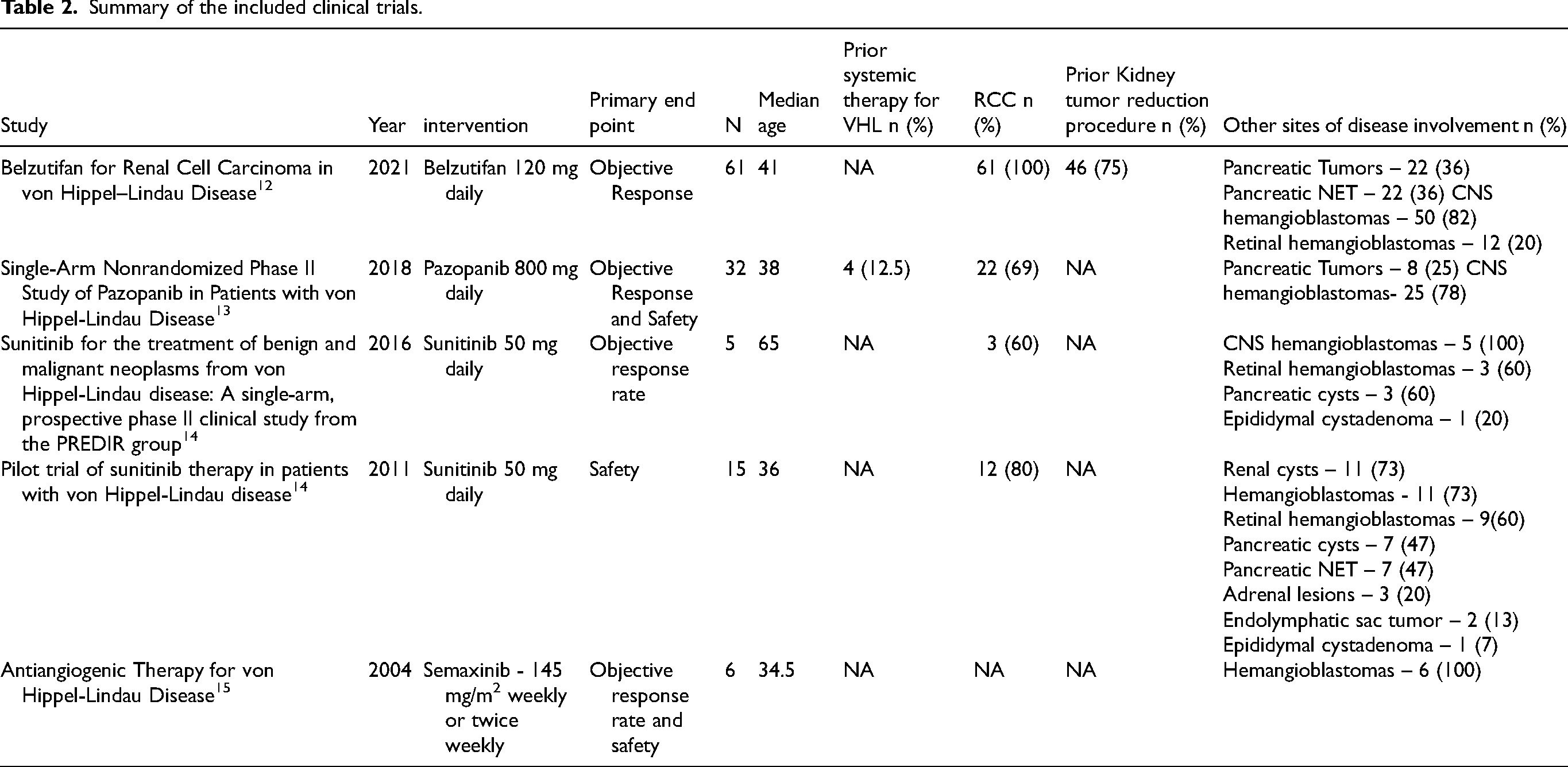
A total of 64 unique citations were identified on the initial search, all of which were screened. Five studies met the inclusion criteria. All five studies were phase 2, single-arm clinical trials that assessed systemic treatments in patients with von Hippel-Lindau Disease related neoplasms. The publication dates of these studies ranged from 2011 to 2018. The assessed therapeutic strategies were three tyrosine kinase inhibitors (TKI) and one hypoxia-inducible factor 2α (HIF-2α) inhibitor (Table 2).
Summary of the included clinical trials.
Semaxinib
Semaxinib is a potent inhibitor of the Flk-1/KDR VEGF receptor tyrosine kinase, targeting the VEGF pathway, inhibiting the VEGF receptor-2, therefore inhibiting angiogenesis. 16 The phase 1/2 study by Madhusudan et al. included six patients with progressive VHL treated with semaxinib. These patients received semaxinib 145 mg/m2 weekly or twice per week. The study's primary endpoints were overall response rate (ORR) and safety. Only results on hemangioblastomas were reported in the study included. Thirty-three percent (33%) of patients experienced disease stabilisation. Furthermore, the study also commented on decreased VEGF levels after treatment initiation, although this did not correlate with disease response. 15
The most commonly seen adverse events with this therapeutic strategy were headaches, nausea/vomiting, fatigue, diarrhoea, arthralgia, and pruritus. 15
Sunitinib
Two of the included studies investigated the VEGF receptor-targeted agent sunitinib in patients with VHL. Sunitinib is another multitargeted TKI. This molecule has documented activity on VEGFR-1, VEGFR-2, and other tyrosine kinase receptors such as KIT, PDGFR-α, and PDGFR-β, ultimately leading to inhibition of endothelial cell proliferation, tumor growth, and pericyte proliferation. 17
The first study by Jonash et al. enrolled 15 patients with VHL. All patients received sunitinib at a dose of 50 mg daily for 28 days, followed by a 14-day break for up to 3 cycles. Patients who experienced grade 3 or higher adverse events had treatment withheld; treatment was restarted at a dose of 37.5 mg when the adverse event decreased to grade 1 or better. The study's primary endpoint was safety, and the secondary endpoint was efficacy by assessing the rate of complete and partial radiographic responses. 18
In this study, 6 out of the 18 RCC lesions showed a partial response (p = 0.014), while 10% of lesions showed progressive disease while on treatment. The mean size decrease by the end of the fourth cycle of sunitinib was 14.4%. All patients with documented PNET showed stable disease with a mean reduction of 12.7% from baseline measurement. On the other hand, none of the patients with retinal angiomas showed a decrease in tumor size. 18
The second study by Oudard et al. enrolled five patients with confirmed VHL disease who were not candidates for further focal treatment. All patients received sunitinib at a dose of 50 mg daily for 28 days, followed by a 14-day rest. The study's primary endpoint was ORR, while the secondary endpoints included tolerability and overall survival. 14
In total, five patients were enrolled, three of whom had RCC. All patients had hemangioblastomas. Three patients (60%) experienced a partial response, one of which was long-lasting. However, no data were available on the specific response for the RCC lesions. All three patients who experienced a response had hemangioblastomas, one with bilateral retinal hemangioblastomas and two with CNS hemangioblastomas. 14
Fatigue, mucositis, diarrhoea, and nausea were the most common adverse events seen in patients treated with sunitinib in both studies. Most of these events were grade 1–2. The most commonly observed grade 3 events were liver enzyme elevation, fatigue, and proteinuria.14,18
Pazopanib
One study assessed pazopanib in patients with clinical manifestations of VHL. Pazopanib is a TKI that targets VEGF, PDGF, cKIT, and Flt-3 receptors, ultimately inhibiting angiogenesis and tumor growth. 19
This study enrolled 32 patients with genetically confirmed VHL or a strong family history and clinical manifestations of VHL. The primary endpoints included ORR and safety. The secondary endpoints were the growth rate over time and the need for surgical intervention. Patients were treated with pazopanib 800 mg daily for 24 weeks. After completing this, patients were allowed to continue therapy. 13
The median follow-up of patients with RCC treated with pazopanib was 12 months (IQR 7–32). Of the 22 patients with documented RCC, 1 (4%) experienced a complete response, while 13 (59%) and 8 (36%) showed a partial response or stable disease, respectively. None of the patients with documented RCC experienced primary disease progression. Importantly, none of the patients enrolled in the study experienced metastatic RCC. 13
Twenty-five patients with CNS hemangioblastomas were included in the study. Of these, 24 (96%) experienced stable disease, and 1 (4%) experienced a partial response. Similarly, of the eight patients with pancreatic tumors, all derived clinical benefit from pazopanib therapy, with 5 (62%) experiencing a partial response and 3 (38%) having stable disease. 13
The most common adverse event encountered with pazopanib was diarrhoea, experienced by 72% of patients, all of which were grade 1 or 2. This was followed by fatigue (69%), liver enzyme elevation (59%), and skin hyperpigmentation (56%). Of the 31 evaluable patients, 10 (32%) remained on the maximum dose of pazopanib (800 mg), while 12 patients (39%) and six patients (19%) had dose reductions to 600 mg and 400 mg, respectively. At the time of final follow-up, only seven patients (23%) remained on therapy. Seven patients (23%) discontinued treatment because of toxicities, 6 (20%) due to progression, 11 (35%) due to patient preferences, and 1 (3%) passed away during the study. 13
Bezultifan
The included study on belzutifan enrolled a total of 61 patients with VHL disease and RCC. Belzutifan is a second-generation HIF-2α inhibitor; this inhibition leads to tumor growth inhibition by decreasing the expression of VEGF, cyclin D1, glucose transporter 1, and EPO. 12 In VHL associated RCC, HIF transcription factor overactivation leads to tumorigenesis through tissue hypervascularization. 20 This HIF dependency serves as the biological explanation of belzutifan efficacy in VHL related tumors.
Patients received 120 mg of Belzutifan daily. The primary endpoint was objective response, specifically in the RCC tumors, and secondary endpoints included duration of response, time to response, and progression-free survival. 12
At a median follow of 21.8 months, belzutifan therapy had an objective response rate of 49% (95% confidence interval [CI], 36–62). Furthermore, 49% of patients had stable disease as their best response for a clinical benefit rate of 97%, as only 3% of patients experienced disease progression while on belzutifan. The progression-free survival rate at 24 months was 96% (95% CI 87–99). 12
Belzutifan therapy also led to significant disease control in non-RCC tumors. For instance, 77% of patients with pancreatic tumors experienced a response, 10% of which experienced a complete response. A significant proportion of patients with PNET (91%) experienced a response, including 14% of which experienced a complete response. Similarly, 30% of patients with CNS hemangioblastoma had a confirmed response, of which 5% experienced a complete response. 12
In an updated analysis at a median follow-up of 37.8 months, in patients with RCC, belzutifan therapy was associated with an ORR of 64% (95% CI 50.6–75.8), of which 90% were partial responses and 10% were complete responses. Similarly, for CNS hemangioblastoma, the ORR was 44% (95% CI 30.0–58.7), with most patients (82%) experiencing a partial response. 21
Belzutifan therapy was overall well tolerated, with only 15% of patients requiring dose modification. The most commonly seen adverse events were anaemia, fatigue, headaches, and dizziness. Most of these adverse events were grade 1 or 2, with only 33% of patients experiencing grade 3 events and only 2% of patients having grade 4 events. As previously observed, all patients treated with belzutifan experienced a reduction in their baseline hemoglobin level during the initial 13 weeks of therapy. This is an on-target, expected toxicity due to the inhibition of the EPO gene, which leads to a reduction in EPO production and, therefore, decreased erythropoiesis. However, only 7% of patients required blood transfusions. 12 The use of erythropoietin-stimulating agents (ESA) appeared to be safe in this population and was used in 14 (23%) patients. The use of ESA was not associated with worse clinical outcomes such as reduced response rate or drug exposure. 22 Only one patient experienced transient hypoxia. This resolved after holding the medication for one week, followed by a dose reduction to 80 mg. 12
Discussion
Although survival among patients with VHL has improved over time, these patients continue to have excess mortality compared to the general population. 6 Currently, approximately 80% of deaths in patients with VHL are VHL-related, with CNS hemangioblastomas and RCC accounting for most of these events. 3 At the present time, roughly a third of VHL deaths are related to RCC. 6 However, patients with RCC only represent a fraction of patients included in most VHL clinical trials, with the exception of the recent belzutifan clinical trial,12,21,23 as such, there is an unmet need to develop more RCC-focused clinical trials in the VHL population, as this continues to be one of the leading causes of mortality in this patient population.
Understanding the biological process described above has opened the opportunity for pharmaceutical approaches for patients with VHL syndrome with the overarching goal of improving outcomes. The most novel therapeutic intervention in this setting is the development of the HIF-2α inhibitor belzutifan. Two phase-one clinical trials investigated belzutifan in patients with previously treated advanced sporadic clear cell RCC (ccRCC) and established the safety and efficacy of this intervention.23,24
In the phase 2 clinical trial included in our systematic review, which led to the approval of belzutifan for the treatment of VHL disease, the clinical benefit of this intervention was not only limited to RCC, but also patients with the other commonly seen VHL-associated neoplasms, such as pancreatic lesions and CNS hemangioblastomas, also derived clinical benefit from the intervention. Among the most clinically important benefits of this intervention is the decrease in surgical procedures needed by patients. Before initiating therapy, 327 procedures had been performed in this population, of which 20% had occurred in the 2.5 years preceding treatment initiation. On the contrary, only three tumor reduction surgeries had occurred following treatment initiation. 12 Given that surgical interventions are a significant source of morbidity and mortality in patients with VHL, delaying surgical interventions is of utmost importance in this population.
In VHL disease, two phase 2 clinical trials have investigated the role of sunitinb therapy. With the caveat of a small sample size, these studies showed clinical benefit from using sunitinib, not only within RCC but also in non-renal manifestations of VHL disease, such as NET and CNS hemangioblastomas. This therapy was associated with significant adverse events such as grade 3/4 fatigue, diarrhoea, stomatitis/mucositis, and hypertension.14,18
Similarly to sunitinib, pazopanib is a multi-target TKI with activity on VEGFR and PDGFR. The effects of pazopanib on metastatic RCC were first assessed by a phase II clinical trial, which showed an ORR of 35% (95% CI 28.4–40.9) and a median PFS of 52 weeks. Importantly, in 45% of patients treated with pazopanib, the best-evidenced response was stable disease. 25 In patients with VHL, pazopanib showed an ORR of 42% (all partial responses), and 58% exhibited stable disease. Complete responses were only seen in RCC lesions; of 59 target renal lesions, 3% experienced complete responses. Although no other site of disease involvement showed complete responses, 53% of patients with pancreatic lesions and 4% of patients with hemangioblastomas had documented responses. It is, however, important to mention that no patients developed new lesions while on pazopanib therapy. 13
Ultimately, when compared with VEGF-TKI therapy such as sunitinib and pazopanib, treatment with belzutifan was not only associated with a better safety profile, with most patients being able to tolerate the recommended dosing without dose modification but also with better responses in both RCC and non-RCC lesions and a decreased rate of surgical interventions directed at VHL-related neoplasms. Given this superior safety profile and response rate, belzutifan is the preferred therapeutic strategy for VHL patients.
This study has several limitations. First, as a systematic review, the strength of our observation is directly associated to the strength of the described studies, all of which were small phase-2-non-randomised clinical trials. Similarly, the original intention of our review was to assess the different pharmacologic strategies for VHL associated RCC, which was not possible given that most VHL studies only included a fraction of RCC patients.
Conclusion
The increasing understanding of the molecular basis of VHL has led to the investigation of several pharmacological therapeutic interventions. However, only a handful of clinical trials have been carried out, all phase-2 non-randomised clinical trials. This is understandable given the rarity of this condition, making the execution of a phase-3 clinical trial not feasible. Among these therapeutic strategies, two VEGF-TKI (sunitinib and pazopanib) have been shown to be effective for treating VHL, with a significant proportion of patients deriving clinical benefit from this strategy. However, these were limited by significant toxicity, which is a characteristic of these pharmaceutical interventions. Belzutifan, on the other hand, has a better toxicity profile and, more importantly, efficacy. However, there continues to be an unmet need for the identification and development of other therapeutic approaches for the treatment of VHL syndrome and its associated malignancy, given that these continue to be a significant source of mortality in patients with VHL. In the meantime, we recommend widespread utilisation of belzutifan in patients with VHL to delay surgical interventions, ultimately decreasing morbidity and mortality in this population.
Footnotes
Acknowledgements
The authors have no acknowledgments.
Author contributions
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study has been supported by the Huntsman Cancer Institute Translational Scholar Grant under Award Number P30CA042014.
Declaration of conflicting interests
Data availability
The data supporting the findings of this study are available within the article and/or its supplementary material.