
Abstract
Clear cell renal cell carcinoma (ccRCC) is the most common malignant neoplasm of the kidney and belongs to the few human tumors known to develop from mutations of the VHL tumor suppressor gene. VHL germline mutations are associated with hereditary ccRCCs in VHL disease. However, somatic VHL gene defects may also occur in sporadic ccRCCs. In this study, we analyzed the frequency and the spectrum of VHL gene alterations in 35 Italian patients with sporadic renal cell carcinoma (RCC). Tumor-specific intragenic VHL pathogenic mutations were detected in 38% (11/29) of the ccRCC patients and 33% (2/6) of the patients with other types of RCC. One novel 18-bp in-tandem duplication and 4 previously unreported nucleotide changes in the VHL gene were described. Microsatellite analysis showed loss of heterozygosity for at least 1 informative marker in 43% (9/21) of the ccRCCs and 50% (3/6) of the non-ccRCCs; 5 of the 13 tumors (38%) harboring VHL gene alterations also had loss of heterozygosity for at least 1 microsatellite marker. Our results confirm that somatic inactivation of the VHL gene may play a pivotal role in the tumorigenesis of sporadic ccRCCs in Italian patients and suggests that mutation analysis of the VHL gene may be helpful for discriminating sporadic, VHL-gene-related ccRCCs from those related to VHL disease.
Introduction
Renal cell carcinoma (RCC) is a rare tumor and accounts for 2%-3% of all malignant diseases in adults (1). Clear cell renal cell carcinoma (ccRCC) (2) is the most common malignant neoplasm of the kidney, is usually a sporadic isolated tumor and belongs to the few human tumors known to develop from mutations of the Von Hippel-Lindau (VHL) tumor suppressor gene.
The human VHL gene (NM_000551) is a tumor suppressor located on the short arm of chromosome 3 (position 3p25.3) (3) and is widely expressed in both fetal and adult tissues (4). The VHL gene encodes 2 protein isoforms of 213 and 160 aminoacid residues, with a predominant cytoplasmic localization (5). One of the main functions of the VHL gene product is its involvement in hypoxia-induced angiogenesis through the formation of a multiprotein complex with elongin B, elongin C, and Cullin 2 (VCBCUL2 complex) (6-9). This complex is best known for its ability to target hypoxia-inducible factors (HIF) for polyubiquitination and proteasomal degradation of several cellular proteins. Moreover, HIF-independent pVHL functions have also been described as being relevant to tumor development, including maintenance of the primary cilium, regulation of extracellular matrix formation and turnover, and modulation of cell death in certain types of cells (due to growth factor withdrawal or in response to other forms of stress) (10-15). Germline mutations in the VHL gene lead to the development of the VHL disease (MIM# 193300), a rare dominantly inherited familial cancer syndrome with a marked phenotypic variability predisposing to central nervous system (CNS) and retinal hemangioblastomas, as well as to ccRCC and pheochromocytomas (16-19). VHL somatic mutations have also been related to the pathogenesis of sporadic ccRCCs in 20%-82% of cases in the United States, Europe and Japan (20-24) and in 8%-20% of cases in a Dutch study of ccRCCs (25). In the present study, we performed molecular analysis of the VHL gene on 35 tumor tissues of Italian patients affected by sporadic RCCs. Tumor-specific intragenic VHL mutations were detected in 11 out of 29 ccRCC patients (38%) and in 2 of the 6 (33%) non-ccRCC patients, confirming the involvement of somatic inactivation of the VHL gene in the tumorigenesis of sporadic ccRCCs in a sample of the Italian population.
Materials and Methods
Patients and Tissue Specimens
A total of 35 patients from Southern Italy with isolated RCC were recruited at the Department of Biomedical Sciences of the University of Foggia (Italy) between 2004 and 2006. For all cases, clinicopathological information including sex, age, tumor type, age at diagnosis, stage, and lymph node status were collected (Tab. I). Since the aim of this study was to evaluate the actual incidence of VHL somatic mutations in sporadic forms of RCC, patients with a family history of renal cancer or with a history of another malignancy were excluded. Following the revision and approval of the study by the local ethical committee, written and informed consent was obtained from each patient before enrollment. Peripheral blood and tissue samples were collected from all patients before and after surgical treatment. A dedicated pathologist reviewed all specimens, using the TNM 2009 for tumor staging. All tumor samples and paired tumor-free normal tissues were formalin-fixed and paraffin-embedded. Specifically, we collected 29 ccRCCs, 5 non-ccRCCs (2 papillary renal cell carcinomas [pRCC] and 3 chromofobe renal cell carcinomas [cRCC]) and 1 tumor tissue composed of both ccRCC and cRCC.
Pathological Characteristics Of The Rcc Patients’ Cohort

DNA was extracted from paraffin-embedded tissues as previously described (26). Briefly, both normal and tumor tissue from unstained paraffin sections were reviewed by the pathologist; each block with tumor tissue was carefully microdissected to ensure that it contained more than 70% of tumor cells. Normal renal tissue was used as source of control DNA for each patient in loss of heterozygosity (LOH) experiments. It was previously reported that artifacts may occur when using archived paraffined material and that the chance of introducing these artificial mutations in formalin-fixed material was inversely correlated to the number of cells used for PCR (27). However, the number of cells in our analyses was large (>500 cells), since we used 10-μ slices of paraffin-embedded material.
DNA from peripheral blood lymphocyte (PBL) samples was extracted using the standard phenol-chloroform procedure (28). DNA concentration was quantified by measuring its absorbance at 260 nm and 280 nm, using a Nanodrop spectrophotometer.
Molecular genetic analysis
Mutation analysis
PCR amplification was performed for the entire VHL coding sequence and the promoter region, including exon-intron boundaries. The set of primers was original and is shown in Table II. PCR products were purified, using GFX™ PCR DNA and Gel Band Purification Kit (GE Healthcare, Buckinghamshire, UK), and sequenced. Sequencing reactions were loaded on an ABI 3100 capillary sequencer (Applied Biosystems, Foster City, CA) and analyzed using the Sequencing Analysis software v.3.7 (PE Applied Biosystems). Once mutations in the DNA from tumor samples were detected, we searched for the same mutations in the corresponding PBL, as to identify germline mutations.
Primer Sequences for VHL Gene Amplification and LOH Analysis

The 18-bp duplication in exon 2 was characterized by cloning the PCR products from mutated samples into StrataClone™ PCR Cloning Vector pSC-A (Stratagene), amplifying the products directly from crude lysates of single bacterial colonies, and direct sequencing.
VHL gene deletions analysis
Analysis of genomic deletions in the VHL gene on DNA extracted from patients’ PBL was performed in long PCR by using a set of primer pairs previously described (29). Briefly, large fragments were amplified in a final volume of 25 μL, using 2U of Expand Long Template Enzyme (Roche), 20 pmol of each primer, 500 μM dNTPs, and 400 ng of template DNA.
Loss of heterozygosity (LOH) analysis
Fluorescent LOH analysis using genomic DNA from matched peripheral blood samples and tumor tissues was performed using 2 microsatellite markers flanking the VHL gene: D3S1335 and D3S1304. PCR was performed in a 50 μL reaction volume containing 2.5 μL of 10X PCR Buffer (Eppendorf), 0.25 nM dNTPs, 20 pmol of each primer, 1 U HotMaster Taq (Eppendorf) and 100 ng of DNA. Cycling conditions consisted of 40 cycles of the LMS2 (ABI Prism™ Linkage Mapping Set version 2) method, performed on a GeneAmp PCR System 9700 (Applied Biosystems) for all markers.
After capillary array electrophoresis (ABI 3100, Applied Biosystems) and data analysis by the ABI Genescan and Genotyper Software 3.7 (Applied Biosystems), the LOH or allelic imbalance (AI) value was quantified using the following formula: (peak 1 height/peak 2 height in tumor DNA)/(peak 1 height/peak 2 height in normal DNA). For this calculation we followed more strict criteria than those indicated in a previous paper (30). In the present study, the cutoff values for defining a LOH were: <0.65 or >1.5. LOH was confirmed at least twice for each marker.
Methylation-specific PCR analysis
DNA extracted from tumor samples was subjected to bisulphite treatment and DNA purification using the Epitect Bisulfite kit (Qiagen Sci, MD USA) according to the manufacturer's instructions. Bisulphite-modified DNA was used as template for conventional methylation-specific PCR. Conventional methylation-specific PCR was carried out as previously described (31), by using the following primer pairs: U: GTTGGAGGATTTTTTTGTGTATGT (sense) and CCCAAACCAAACACCACAAA (antisense); M: TGGAGGATTTTTTTGCGTACGC (sense) and GAACCGAACGCCGCGAA (antisense) (32). A PCR reaction for the ACTB gene promoter region not containing CpGs was also performed as control of the bisulfite conversion. PCR conditions were the following: 35 cycles at 95°C for 1 minute, 64°C and 72°C for 1 minute. For each PCR reaction CpGenome Universal Methylated DNA (Serological Corp., Norcross GA) was used as positive control, and Universal Unmethylated DNA (Serological Corp., Norcross, GA) was used as negative control. PCR products were run on 3% agarose gel stained with ethidium bromide, and visualized at UV light.
Statistical methods
Associations between tumors pathological parameters and RCC mutational status were assessed by using the chi-square or Fisher tests. All analysis were 2-tailed, and p values <0.05 were considered statistically significant. Statistical analysis was performed with the SPSS 13.0 statistical software package.
Results
Study cohort
Thirty-six patients (18 males and 17 females) with a single isolated RCC and no evidence of VHL disease were investigated. Patients had a mean age of 67 years (range 58-75 years). Distribution of the patients according to the frequency of tumor cell histotypes and TNM stage is shown in Table I. Histological analysis of the surgical specimens showed a predominance of ccRCCs (29 cases; 80%).
Molecular findings
A total of 17 nucleotide changes in the VHL gene were detected in the 35 tumor tissues from RCC patients (46%), with 1 patient presenting 2 different coding mutations in the same sample. Tumor-specific intragenic VHL mutations were detected in 38% (11/29) of the ccRCC patients. One of these ccRCC-affected patients had a germline mutation, since the mutation was observed in both normal renal specimens and peripheral blood. No other germline mutations or large VHL gene deletions were observed in peripheral blood of patients. Most of the mutations were identified in ccRCC patients (15/17; 88%); the other 2 point mutations were identified in 1 case of cRCC and 1 case of mixed tumor type (cRCC and ccRCC). We did not find any significant correlation between the presence of mutations and several clinicopathological variables.
Two different types of mutation were detected in the coding VHL region: point mutations (16/17), and one 18-bp duplication, all in heterozygous state. Only 1 (H125H) of the point mutations identified in the VHL exons was a silent variant, here reported for the first time. The mutations found on codons 33, 73 and 120 (p.S33X and p.Q73X and p.R120X) and the splicing mutations (c.463+2 T>C) resulted in a stop codon and a truncated protein. One 18-bp in-tandem duplication was also found in the VHL gene (c.340_358 dup18), likely affecting protein function by in-frame insertion of 6 consecutive aminoacids (Tab. III). The remaining 8 mutations are single-base substitutions resulting in aminoacidic changes (p.G39D, p.P86L, p.W88C, p.L89H, p.S111N, p.F119L, p.V130F, p.R161G) (Tab. II). Mutations were detected mainly in the exon 1 of the VHL gene (7/13). One of the mutations, a heterozygous 18-bp duplication in exon 2, has not been previously described (Fig. 1). DNA obtained from peripheral blood was available for all patients but only patient RCC-23, with a p.P25L alteration, showed in the germline the same aminoacidic change detected in tumor tissue. However, this variant has been previously described (33-35) and was not considered a mutation, but rather a polymorphism. The design of the primers here used allowed amplification and analysis of the 5'UTR and promoter regions of the VHL gene in the same electropherogram. Two point mutations were identified (c.1-28G>A and c.1-97T>C, rs34271731) in the 5'UTR and VHL promoter regions (Genbank accession No AF010238). In summary, 13 of the 17 mutations (all identified missense, non sense and frameshift mutations) detected in the VHL gene were considered to be serious and all were detected at somatic level.
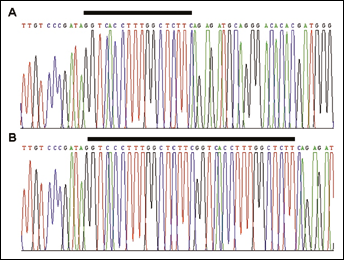
18-bp in tandem duplication of the VHL gene identified in a sporadic RCC case. Representative sequence electropherogram illustrating the sequence of VHL exon 2 region in the wild type (A) and mutated allele (B). The 18-bp duplicated sequence is marked by the bar drawn below the sequence.
Molecular Vhl Gene Alterations Detected In Renal Cell Carcinomas
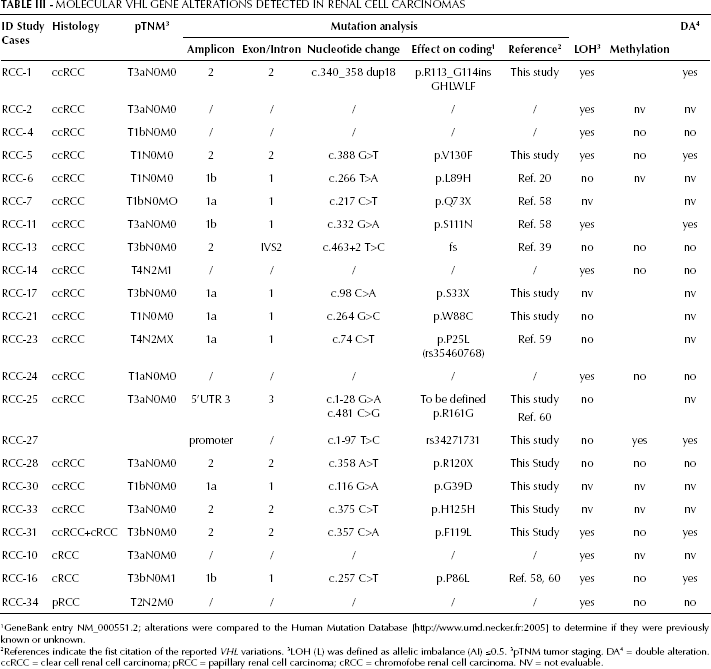
GeneBank entry NM_000551.2; alterations were compared to the Human Mutation Database [http://www.umd.necker.fr:2005] to determine if they were previously known or unknown.
References indicate the fist citation of the reported VHL variations. 3 LOH (L) was defined as allelic imbalance (AI) ≤0.5. 3 pTNM tumor staging. DA 4 = double alteration. ccRCC = clear cell renal cell carcinoma; pRCC = papillary renal cell carcinoma; cRCC = chromofobe renal cell carcinoma. NV = not evaluable.
The results of the LOH analysis are summarized in Table III. Allelic losses on chromosome 3p were found for at least 1 informative marker in 11 out 24 (46%) RCC tumors, mainly ccRCCs (Fig. 2). Finally, 5 of the tumors harboring VHL gene mutations also had LOH for at least 1 microsatellite marker, in agreement with the “two-hit” hypothesis for the inactivation of tumor suppressor genes (36).
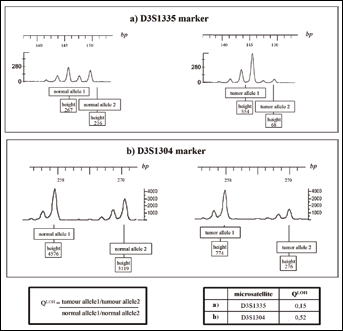
LOH of D3S1335 and D3S1304 microsatellite markers in RCC-2 and RCC-31 samples. The peak heights are measured in relative fluorescence units. (a) for RCC-2: QLOH = (267/1216)/(554/68) = 0.15; (b) for RCC-31: QLOH = (4576/3119)/(774/276) = 0.52.
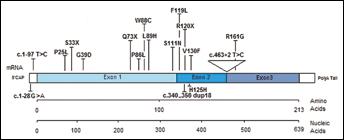
The aminoacidic changes are reported across the structure of the pVHL protein: the 3 exons and the UTR regions are indicated.
Methylation analysis by methylation-specific PCR was performed on 15 cases for which there was enough formalin-fixed, paraffin-embedded material available. Hypermethylation of the VHL gene promoter region was detected only in 1 of the analyzed samples. The lack of promoter hypermethylation was indicated by: (i) failure of amplification of the promoter fragment in all tumor samples and negative controls (Universal Unmethylated DNA, Serological Corp., Norcross, GA) that were tested with the specific primer pair; (ii) amplification of the positive control of PCR reaction CpGenome Universal Methylated DNA (Serological Corp., Norcross GA). ACTB control fragment was amplified correctly in all bisulphite treated DNA, positive and negative DNA controls.
VHL status by patient and tumor features
VHL alterations were stratified by tumor histopathology and patient features, as summarized in Table III. No significant correlations were found between the presence of mutations and the tumors’ or patients’ features examined.
Discussion
The VHL gene is considered a master tumor suppressor gene involved in cell cycle regulation, modulation of hypoxia-inducible genes and proper fibronectin assembly in extracellular matrix (37). Molecular studies examining tumor DNA from sporadic cases of RCC have provided strong evidence that VHL alteration is a common early event in the carcinogenesis process. It has been estimated that approximately 50%-70% of all sporadic ccRCCs harbor biallelic VHL defects (24, 25, 34, 35, 38, 39).
We examined VHL gene alterations using mutational analysis, LOH analysis and determination of promoter methylation status in a cohort of 35 Italian RCC patients. Sixty-eight percent of these patients were found to have at least 1 molecular alteration in the VHL gene (nucleotidic change, methylation or LOH). A total of 17 nucleotidic changes were identified in RCC tumor tissues. One novel mutation (c.340_358 dup18), located within exon 2 of the VHL gene and heavily affecting protein function, was identified in this study in a ccRCC sample. Moreover, 2 previously unreported nucleotidic changes (p.S33X and p.G39D) at the 5’ of codon 54 were identified in 2 different cases of ccRCCs. These changes are unusual and would be expected to affect only the pVHL30 translation product at a specific region, which is known to interact with fibronectin and was recently described as an important mediator for tumor invasion (40-42). In addition, other 12 mutations (8 missense, 3 nonsense and 1 splicing mutation), affecting mainly the first exon of the gene and located into the interaction sites of pVHL with HIF-1α, are functionally related to the development of the disease (43). To note, a double inactivation (a missense mutation and a LOH) were found in a cRCCs case (RCC-16), confirming the recently reported involvement of the VHL gene alterations in the pathogenesis of this type of renal tumor (44, 45).
Several studies have previously examined VHL alterations in different patient populations and have reported significant differences in the prevalence of these mutations, which ranged from 42% to 87% in frozen samples (34, 38, 46, 47) and from 20% to 61% in formalin-fixed, paraffin-embedded tissues (35, 38, 48-52). This is the first study analyzing the VHL genetic alterations in an Italian cohort of sporadic ccRCC cases that reports an incidence of VHL genetic alterations consistent with the lower limit of the range reported in other geographical areas. The slight difference in mutation prevalence could be due to several factors, including the limit of detection of mutant alleles using Sanger sequencing analysis (approximately 15%-20%), the patients’ population examined, the type of material analyzed (archived paraffin material instead of fresh material) and tumor histopathology (53-55). In addition, LOH analysis was performed on all RCC samples, and allelic loss of the VHL gene region was demonstrated in 46% (11/24) of the cases. The existing discrepancy between this latter finding and the previously published data on LOH frequency (which report values ranging from 78% to 93%) is probably due to the more strict criteria used to discriminate loss and retention of VHL alleles than those more frequently used (cutoff value for defining a LOH was lowered to 0.65) (35, 40, 47). Interestingly, 5 cases harboring VHL mutations also showed allelic loss at the VHL locus, suggesting that both alleles are inactivated. Inactivation of the VHL gene can also occur by promoter hypermethylation (36, 53, 57). Due to the low amounts of DNA obtained from formalin-fixed, paraffin-embedded specimens, we could only analyze 15 cases, of which only one (1/15; 7%) showed promoter hypermethylation. This is not surprising if we consider that the previously reported prevalence was of 5%-17% (13, 15, 35, 52).
Finally, we examined possible correlations between our genetic findings and patients’ clinical and descriptive features. The prevalence of mutations was not associated with any of the parameters examined in terms of tumors’ or patients’ features. In accordance with previous reports, our findings confirm that functional alterations of the VHL gene may also play a pivotal role in RCC tumorigenesis, especially in cases of ccRCCs and, to a more limited degree, in cases of cRCCs. Our findings further support the concept that mutation analysis of the VHL gene is of striking importance for discriminating the sporadic forms of cancer from those related to VHL disease.
Footnotes
Acknowledgements
The authors thank Chiara Di Giorgio (translator and medical writer, BioAgromed, University of Foggia) for linguistic revision.