
Abstract
Rationale:
Central retinal artery occlusion (CRAO) is an ophthalmologic emergency that, without prompt reperfusion, is associated with a high risk of permanent blindness. There is currently no evidence-based, effective treatment for CRAO.
Methods and design:
TenCRAOS is an investigator-initiated, international, multicenter, randomized controlled, double-dummy, double-blind, phase III trial testing tenecteplase (TNK) 0.25 mg/kg + placebo versus acetylsalicylic acid (ASA) 300 mg + placebo. The target population is patients diagnosed with CRAO and a best-corrected visual acuity (BCVA) of ⩾1.0 logarithm of the minimum angle of resolution (logMAR), corresponding to a decimal BCVA of ⩽0.1 or a fraction BCVA of ⩽6/60, who can be treated within 4.5 h.
Sample size:
Assuming a difference in proportion of participants reaching the primary endpoint of 20% in the placebo/ASA treatment group versus 50% in the TNK/placebo group, we need 78 participants to reach 80% power to detect the difference on a 5% significance level.
Outcomes:
The primary outcome is the proportion of participants with BCVA of ⩽0.7 logMAR in the affected eye at 30-day follow-up, corresponding to a decimal BCVA of ⩾0.2 or fraction BCVA of ⩾6/30, representing a clinically relevant improvement of BCVA of ⩾0.3 logMAR (or 15 letters). Secondary efficacy outcomes include proportion of participants with BCVA of ⩽0.5 logMAR, corresponding to a decimal BCVA of ⩾0.32 or fraction BCVA of ⩾ 6/19, self-reported vision-targeted health status, quality of life, and modified Rankin Score at 30 and 90 days. Safety outcomes include symptomatic intracranial hemorrhage, major bleeding, and mortality. Exploratory analyses include assessment with optical coherence tomography with angiography and transorbital ultrasound.
Discussion:
TenCRAOS intends to assess the efficacy and safety of systemic TNK within 4.5 h of CRAO onset.
Introduction and rationale
Central Retinal Artery Occlusion (CRAO) is an ophthalmologic emergency that bears a high risk of severe visual loss, unless swift reperfusion is achieved. 1 The condition is typically the result of an artery-to-artery embolism from a carotid plaque or cardio-embolism, although a broad spectrum of other types of etiology can occur. 2 Patients present with sudden, painless, and severe monocular vision loss accompanied by an afferent pupillary defect. The diagnosis is often based on history, physical, and funduscopic examination, displaying superficial opacification or whitening of the retina in the posterior pole and a bright red foveal area, the so called “cherry-red-spot sign.” 1 The annual incidence of CRAO is two in 100.000 people, highest in men aged 75–84 years, and is expected to increase with aging population.3,4 The visual consequences of a CRAO can be severely disabling, yet there is no evidence-based treatment option.
Whether prompt reperfusion with thrombolytic agents can improve the outcome of CRAO, as proven in ischemic stroke, remains unresolved. Considering the similarities between acute ischemic stroke and CRAO, and the successful application of systemic thrombolysis in the treatment of acute ischemic stroke, it seems reasonable to apply the same approach to CRAO. However, the differences in vascular anatomy and metabolic characteristics between the retina and the brain preclude a direct extrapolation from stroke literature. 5
A first randomized controlled trial (RCT) to compare conservative medical treatment and local intra-arterial fibrinolysis (LIF), the European Assessment Group for Lysis in the Eye (EAGLE) trial, 6 recruited patients with CRAO within 20 h after symptom onset and found no visual improvement. The mean interval between the first symptoms and therapy was 12.8 h, and retinal infarction was likely to be established at this stage. 7 Similarly, an earlier RCT by Chen et al. also evaluated thrombolysis in CRAO but included patients with broad time windows, leading to high rates of intracranial hemorrhage (ICH). 8 The wide time window and risk of bleeding complications are critical limitations of these historical trials.
Intravenous thrombolysis, administered within 4.5 h or earlier, was found to significantly improve outcome compared to controls in a meta-analysis of observational data from 962 patients, whereas other treatments such as ocular massage, anterior chamber paracentesis, and/or hemodilution was found to worsen clinical outcome. 1 Absolute risk reduction associated with systemic thrombolysis was 32.3%, with a number needed to treat of 4.0 (95% confidence interval (CI), 2.6–6.6). Recently, the French multicenter trial, THrombolysis (Alteplase) in Patients With acutE Central retInal Artery Occlusion (THEIA), tested intravenous alteplase within 4.5 h. This study, which recruited 70 participants from 16 sites, was presented at the World Stroke Congress in 2024 with neutral results (ClinicalTrials.gov Identifier: NCT03197194). The findings from THEIA underscore the ongoing uncertainty regarding the efficacy of systemic thrombolysis in CRAO and highlight the need for further investigation. 9
Tenecteplase (TNK) is a tissue plasminogen activator (tPA) produced by recombinant DNA technology. TNK has demonstrated several pharmacological advantages over alteplase: lower risk of bleeding complications, 14-fold greater fibrin specificity, more rapid onset of action, and longer half-life. Moreover, TNK can be given as an intravenous (IV) bolus over 10 s, making it easier to administer than tPA. 10 Although TNK is well-established reperfusion therapy in acute myocardial infarction and is also recently established treatment for acute ischemic stroke.11 –13 So far, only case reports have been published on the use of tenecteplase for IVT in CRAO, which warrants further investigation. 14
The main aim of the Tenecteplase in Central Retinal Artery Occlusion Study (TenCRAOS) is to assess the efficacy and safety of 0.25 mg/kg systemic TNK administered within 4.5 h of symptom onset.
Methods
Study design
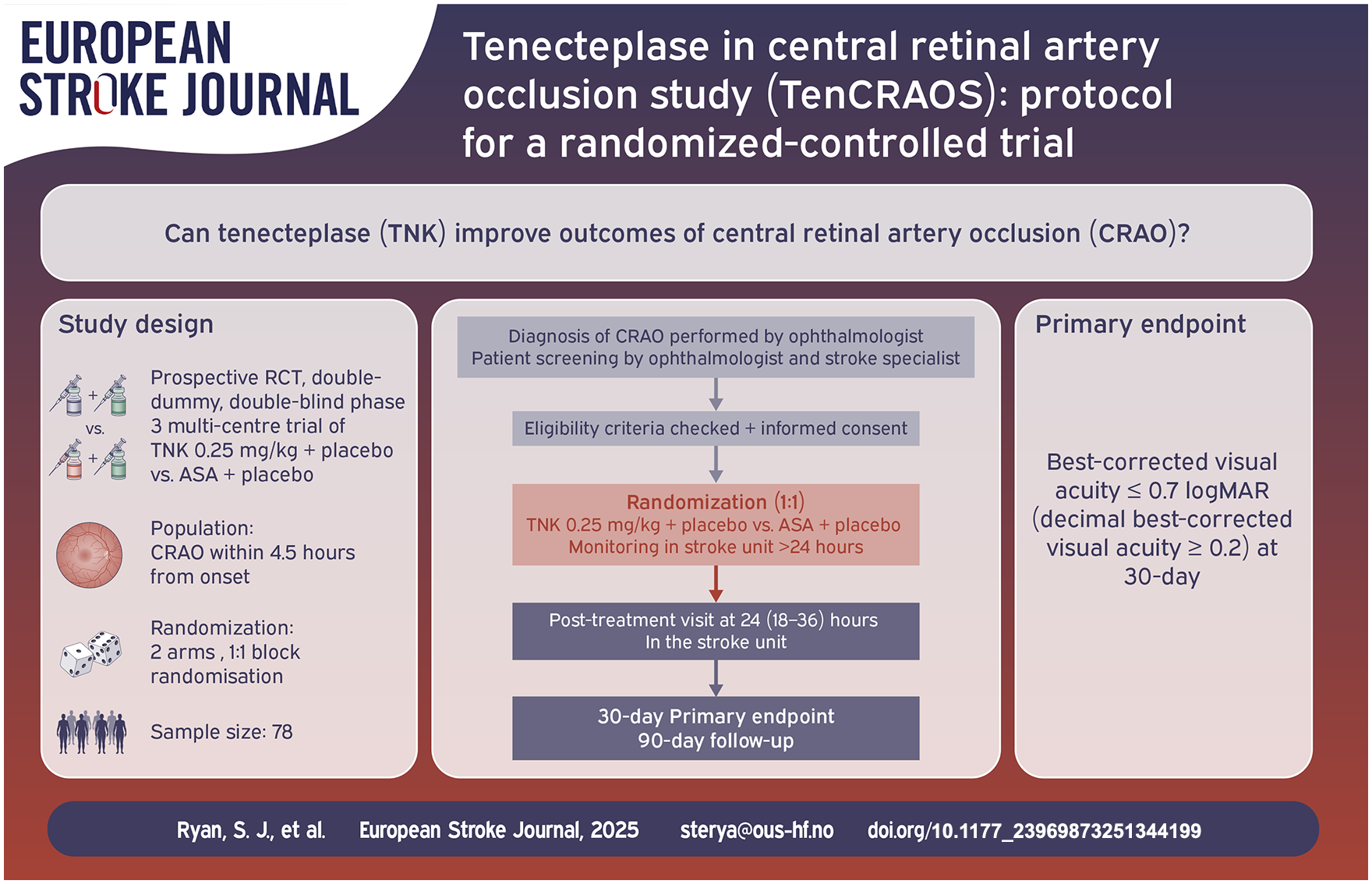
Designed and led by Oslo University Hospital, TenCRAOS is an international, multicenter, prospective, randomized-controlled, double-dummy, double-blind, phase III trial of TNK 0.25 mg/kg + placebo versus ASA + placebo (two arms with 1:1 block randomization) for CRAO within 4.5 h of symptom onset. Maximum dose is 25 mg. At all participating centers, ophthalmologists assert the diagnosis of CRAO by performing an acute eye examination that includes assessment of visual acuity, tonometry, slit lamp examination, and fundoscopy.
Neurological examination and head CT scan is done before inclusion. Blood samples including CRP, hemoglobin, hematocrit, white blood cells, platelet count, natrium, potassium, creatinine, glucose, and INR are collected on admission. However, the results are not required before inclusion except in cases where the participants are on warfarin sodium or there are other potential contraindications requiring this information. In women of childbearing potential, a negative pregnancy test by testing serum or urine human chorionic gonadotropin is done before inclusion if there is a possibility that she is pregnant. Participants are given study medication after written informed consent and managed in a stroke unit where assessment of NIHSS, vital signs and potential adverse events is performed. Control brain imaging with MRI or CT is done after 24 (±6) h.
Etiological investigations are done according to local routines. After treatment in the stroke unit, the participants are re-examined by an ophthalmologist and a stroke physician as an out-patient at (30 ± 5) and 90 (±15) days. The ophthalmologic examinations at the follow-up visits are more extensive and include formal assessment of best-corrected visual acuity (BCVA) with an Early Treatment of Diabetic Retinopathy Study (ETDRS) chart, automated perimetry using the monocular Esterman program, and the National Eye Institute Visual Functioning Questionnaire 25 (NEI-VFQ 25). Please see Figure 1 and Supplemental Table 1.

Trial schedule.
Participant population
TenCRAOS randomizes patients with acute monocular severe vision-loss due to non-arteritic CRAO, verified by an ophthalmologist. Vision must be severely affected to make a complete occlusion probable and support treatment, and the BCVA inclusion criterion of ⩾1.0 logarithm of the minimal angle of resolution (logMAR), corresponding to a decimal BCVA of ⩽0.1 or a fraction BCVA of ⩽ 6/60, also meets the definition of severe visual impairment (or worse) for the affected eye, as defined by the World Health Organization. 15 The treatment must be given within 4.5 h after onset of symptoms. The inclusion and exclusion criteria are listed in Table 1.
Inclusion and exclusion criteria.

logMAR: logarithm of the minimal angel of resolution; IMP: investigational medicinal product; MRI: magnetic resonance imaging; CT: computed tomography; CRAO: central retinal artery occlusion; INR: International Normalized Ratio.
Randomization
Participants are assigned in a 1:1 ratio to one of the treatment arms using block randomization with a computer-generated random allocation sequence. The treatment kits contain either active treatment (TNK as powder with solvent) and dummy co-medication (placebo capsule) or dummy treatment (placebo powder with solvent) and active co-medication (acetylsalicylic acid (ASA) capsule). Each treatment kit is assigned a unique kit number according to the allocation sequence. Once a participant is included, the site personnel authorized for treatment preparation will open and use the treatment kit with the lowest kit number.
Intervention
For this study, IV TNK and IV placebo are defined as Investigational Medicinal Product (IMP). Eligible participants receive capsules with either 300 mg ASA or placebo orally together with the IMP. The dosage of TNK is 0.25 mg/kg body weight, with a maximum dose of 25 mg.
In TenCRAOS we have included an alternate treatment arm consisting of 300 mg ASA to ensure initiation of acute stroke treatment in the absence of IV thrombolysis. This in accordance with international stroke guidelines and proven to be safe and beneficial. 16
Primary outcome
The primary outcome is the proportion of participants with BCVA of ⩽0.7 logMAR, corresponding to a decimal BCVA of ⩾0.2 or fraction BCVA of ⩾ 6/30, in the affected eye at 30 days after treatment. As BCVA of ⩾1.0 logMAR is an inclusion criterion, the primary outcome represents an improvement of BCVA of ⩾0.3 logMAR (or 15 letters).
Secondary outcomes
Multiple secondary outcomes are measured, including proportion of participants with BCVA of ⩽0.7 logMAR in the affected eye at 90 days after treatment, proportion of participants with BCVA of ⩽0.5 logMAR, mean change in BCVA, visual recovery to logMAR ⩽ 0.7, logMAR ⩽ 0.5 in participants who were treated with TNK within 3 h of onset, and monocular Esterman perimetry score at 30 and 90 days. We also measure National Institutes of Health Stroke Scale score (NIHSS) and modified Rankin Scale score (mRS) at discharge, 30 (±5), and 90 days (±15) days. Additionally, two patient-reported outcome measures are utilized at 30 (±5) and 90 (±15) days: EuroQol-5 Dimension (EQ-5D) and National Eye Institute Visual Function Questionnaire (NEI-VFQ 25).
Exploratory outcomes
Two optional exploratory outcomes can be evaluated by all sites: (1) optical coherence tomography (OCT) volume scans and OCT angiography to assess the retinal circulation and (2) point of care ultrasound of the orbit to determine the presence of the “white-spot sign” and measure the optic nerve sheath diameter.
Safety measures
The investigator is responsible for the detection and documentation of events meeting the criteria and definition of an adverse event (AE) or serious adverse event (SAE). The participants are instructed to contact the investigator immediately should they manifest any signs or symptoms they perceive as serious during the follow up period of 90 days.
Assessment of acute complications, such as systemic and intracranial bleeding, is performed by physical examination, measurement of vital signs every hour the first 24 h, and by head CT or MRI after 24 h. If there is clinical suspicion of intracranial hemorrhage earlier than 24 h, head CT or MRI will be performed when appropriate.
Blinding
TenCRAOS is double-blinded. Boehringer Ingelheim (Ingelheim, Germany) provides TNK and intravenous placebo solution, and Kragerø Tablettproduksjon (Kragerø, Norway) provides ASA and placebo capsules to the pharmacy at St. Olav University Hospital in Trondheim, Norway, which labels, packs, and distributes the study medication and unblinding envelopes to the participating trial sites. The unblinding envelopes are only opened if it is medically imperative. The pharmacy and the statistician in the Data Safety Monitoring Board (DSMB) have access to the participant list.
Data monitoring and safety committee (DSMB)
An independent DSMB performed unblinded reviews of SAEs in all participants after the first 20 and 40 participants have completed all study visits. The DSMB adjudicates the occurrence of clinical study safety endpoints in accordance with the definitions specified in the protocol. Planned meetings occur throughout the study.
The DSMB advises the chairperson of the Trial Steering Committee regarding the continuing safety of the participants and those yet to be recruited into the study, and as to the continuing validity and scientific merit of the trial.
Sample size estimation
The sample size estimation is based on recent meta-analyses1,5 and their primary endpoint: proportion of participants with ⩽ 0.7 logMAR BCVA at 30 (±5) days after treatment, representing an improvement in BCVA of ⩾0.3 logMAR (or 15 letters). Assuming a difference in the proportion of participants meeting the primary endpoint of 20% in the placebo/ASA treatment group versus 50% in the TNK/placebo group, we need 78 participants (39 in each group) to reach 80% power to detect the difference on a 5% significance level. There will be no replacement for participants lost to follow-up.
Study conduct
The trial is being conducted at 27 sites in 8 countries in Europe and Australia. The study enrolled the first participant in November 2020, and estimated study completion date is by end of March 2025.
Statistical analyses
Logistic regression will be employed as the primary analysis method, with the primary endpoint as dependent variable, using treatment and center as independent variables. The primary estimator of treatment effect is the difference in the probability of visual recovery to logMAR ⩽ 0.7 between the treatment groups, obtained by applying the delta method to the logistic regression model parameters. The primary analysis will be presented with the number and percentage of participants obtaining visual recovery by treatment group, the estimate of the primary estimator with 95% confidence limits, and the p-value of the primary null hypothesis. The null hypothesis will be evaluated on the 5% significance level. As there is only one primary analysis, there will be no adjustments for multiplicity testing.
Study organization and funding
Oslo University Hospital is the trial sponsor. The main sources of funding are Klinbeforsk (Norwegian Programme for Clinical Therapy Research), the South-Eastern Norway Regional Health Authority, and Odd Fellow. Boehringer-Ingelheim provided tenecteplase and intravenous placebo free of charge as well as financial support to the sites outside Norway but had no influence on the study conduct, analysis, or interpretation. Boehringer-Ingelheim was given the opportunity to review the manuscript for medical and scientific consistency as it relates to Boehringer-Ingelheim substances, as well as intellectual property considerations.
Research ethics and regulatory approvals
The trial is conducted in accordance with the Medical Research Council Guidelines for Good Clinical Practice in Clinical Trials, the Council of Europe’s Convention on Human rights and Biomedicine (CETS No.: 164), the ICH Harmonized Tripartite Guideline for Good Clinical Practice (CPMP/ ICH/135/95), and the Declaration of Helsinki (Edinburgh, 2013). Ethical approval for the trial was granted by the Regional Committee for Medical Research Ethics South East Norway and, subsequently, by local authorities and competent regulatory authorities at all participating sites. Written, informed consent is obtained from all eligible participants according to national regulations.
Discussion
TenCRAOS is an international, randomized controlled trial jointly run by neurologists and ophthalmologists, addressing an important unanswered clinical dilemma: Is acute reperfusion treatment with systemic thrombolysis beneficial and safe in CRAO? Observational studies do suggest that IV thrombolysis is favorable in CRAO. However, the efficacy of intravenous thrombolytic therapy has so far not been proven.1,5,17 The THEIA trial, which is not yet published, was presented at the World Stroke Congress in 2024 with neutral results. 9 The ongoing REVISION trial (ClinicalTrials.gov Identifier: NCT04965038) led from Germany, started with tPA and will also use TNK. The primary outcome of the REVISION trial is functional recovery to normal or mildly impaired vision in the affected eye defined as best-corrected visual acuity of the Logarithm of the Minimum Angle of Resolution of 0.5 or less at 30 days with planned recruitment numbers up to 422 participants (ClinicalTrials.gov Identifier: NCT04965038). 18 In TenCRAOS we have this endpoint as one of the secondary endpoints.
Since the administration of the study medication and the acute management of the participants in these trials are handled by stroke units, the participants are included in an established stroke pathway after their CRAO diagnosis has been verified by ophthalmologists. The use of an established stroke pathway and the pragmatic study design, including therapeutic time window of 4.5 h, facilitate these trials in the acute phase of the disease. The therapeutic window in CRAO might be shorter than in ischemic stroke, as the central retinal artery is mainly an end-artery and only has sparse collaterals from short cilia arteries stemming from the ophthalmic artery. However, the time window is identical to two other RCTs of thrombolysis in CRAO; THEIA and REVISION. 18 Furthermore, the design of our trial is based on collaboration with the other trials, facilitating a later meta-analysis. The results from the three trials could also answer whether the pharmacological differences between alteplase and TNK are clinically relevant in CRAO.
One of the main obstacles in treating acute CRAO promptly is the time taken for patients to be admitted. The current practice, or lack thereof, has established a reduced sense of urgency amongst medical professionals and patients alike. Patients often present many hours or even days after onset. 17 We hope to overcome this obstacle through communication to general practitioners, ophthalmologists, prehospital services, and the public. The practical pathway is a little different from regular stroke patients, as we also include ophthalmologists in the well-established acute-stroke-thrombolysis workflow. All participants must receive a timely diagnosis of CRAO from an ophthalmologist before being considered for inclusion. This was seen as a potential obstacle before initiation of the study, but the experience gained through TenCRAOS so far shows good feasibility for such a pathway.
By defining the primary endpoint as the proportion of participants with BCVA of ⩽0.7 logMAR in the affected eye at 30 days, a relatively low sample size of only 78 participants is sufficient, facilitating the trial. As ⩾ 1.0 logMAR is an inclusion criterion, BCVA of ⩽0.7 logMAR at 30 days represents an improvement in BCVA of ⩾0.3 logMAR (or 15 letters), which is broadly regarded as a clinically significant improvement in BCVA. Moreover, CRAO patients may present with severe visual impairment at the level of light perception, which is not regarded a numerical measurement of visual acuity. 19 Accordingly, calculating change in visual function before and after treatment as a continuous variable will be hampered by uncertainty in CRAO, whereas our study has the advantage of defining the primary endpoint as a categorical variable.
There is also a planned IPD meta-analyses of all three RCTs evaluating thrombolysis within the early therapeutic time window of 4.5 h, where additional outcomes will be investigated. We have taken a conservative approach in TenCRAOS, using a long list of exclusion criteria to reduce the bleeding risk. If the thrombolytic therapy is proven effective and safe in CRAO, we expect the number of criteria to be reduced in the same manner as we have seen in acute ischemic stroke6,8,20
TenCRAOS is an international multi-center study, in which eight countries collaborate in establishing treatment pathways for CRAO in a research setting. In turn, the feasibility of incorporating acute thrombolysis for CRAO into clinical care, utilizing current acute stroke algorithms, will also be proven and can be readily incorporated, should the trial show positive results.
Conclusion
TenCRAOS will answer whether TNK 0.25 mg/kg administered within a 4.5-h time window is an effective and safe treatment for CRAO, using the primary endpoint of proportion of participants with BCVA of ⩽0.7 logMAR in the affected eye at 30 (±5) days after treatment.
Supplemental Material
sj-docx-1-eso-10.1177_23969873251344199 – Supplemental material for Tenecteplase in Central Retinal Artery Occlusion Study (TenCRAOS): Protocol for a randomized-controlled trial
Supplemental material, sj-docx-1-eso-10.1177_23969873251344199 for Tenecteplase in Central Retinal Artery Occlusion Study (TenCRAOS): Protocol for a randomized-controlled trial by Stephen J Ryan, Øystein Kalsnes Jørstad, Mona Skjelland, Claus Z Simonsen, Toke Bek, Rolf Ankerlund Blauenfeldt, Petra Ijäs, Arja Laitinen, Andrej N. Khanevski, Jørgen Krohn, Eyvind Rødahl, Robin Lemmens, Jelle Demeestere, Catherine Cassiman, Ingvild Nakstad, Kristin Evensen, Tiril Sandell, Steffen Hamann, Louisa M Christensen, Sverre Rosenbaum, Vaidas Matijosaitis, Reda Zemaitiene, Hanne Ellekjær, Dordi Austeng, Thomas C Truelsen, Michael V Mazya, Frank Träisk, Pauli Ylikotila, Ulpu Salmi, Kristian N Jenssen, Håvard Lisether, Cathrine Breivik, Kristina Devik, Lasse-Marius Sandes Honningsvåg, Jurgita Valaikienė, Andrius Cimbalas, Vetle Nilsen Malmberg, Espen Anderson, Sylvie De Raedt, Marcel Ten Tusscher, Noémie Ligot, Deborah Lipski, Fredrik Björck, Annelie Hamrin, Tore Solbakken, Ane Roushan Tharaldsen, Anette Huuse Farmen, Andreas Helgesen, Stein Harald Johnsen, Geir Bertelsen, Åse Hagen Morsund, Erik Holen, Arnstein Tveiten, Henrik B Johannessen, Peter Kelly, Evelyn O’Neill, Ansar Roy, Christina Kefaloykos, Thor Håkon Skattør, Kristian L Kraglund, Lauren Sanders, Peter Vanacker, Daniel Strbian, Morten C Moe and Anne Hege Aamodt in European Stroke Journal
Footnotes
Acknowledgements
The authors thank the user organizations “Blindeforbundet” and “LHL Hjerneslag og Afasi” represented by steering committee member Arild Hagen and DSMB (Ole Morten Rønning, Kjell-Arne Kvistad, Martin Ystad and Inge Christoffer Olsen), and TenCRAOS investigators for their guidance, diligence, and support. We thank Boehringer Ingelheim for supplying the tenecteplase and placebo for the trial and for financial support of sites outside Norway.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: A.H. Aamodt has received honoraria for advice or lecturing from Pfizer Allergan, Teva, Novartis, Roche, Lundbeck and Teva and research grant from Boehringer Ingelheim.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Oslo University Hospital is the trial sponsor. The main sources of funding are Klinbeforsk (National program for clinical treatment research in the specialist health service), the South-Eastern Norway Regional Health Authority, and Odd Fellow. Boehringer-Ingelheim provided tenecteplase and intravenous placebo free of charge as well as financial support to the sites outside Norway but had no influence on the study conduct, analysis, or interpretation. Boehringer-Ingelheim was given the opportunity to review the manuscript for medical and scientific consistency as it relates to Boehringer-Ingelheim substances, as well as intellectual property considerations.
Informed consent
Written, informed consent is obtained from all eligible participants, before the study, according to national regulations.
Ethical approval
Ethical approval for the trial was granted by the Regional Committee for Medical Research Ethics Southeast Norway (Ref. 2019/327) and, subsequently, by local authorities and competent regulatory authorities at all participating sites.
Guarantor
Anne Hege Aamodt
Contributorship
The following authors designed the TenCRAOS trial protocol: AHA, MCM, ØJ, ICOKLK, and SJR. Stephen James Ryan drafted the publication of the protocol and all other authors have revised the manuscript and made critical comments.
ORCID iDs
Trial status
As of June 1st, 2025, the inclusion of 78 participants into TenCRAOS was completed.
Trial registration
ClinicalTrials.gov NCT04526951, EudraCT Number 2014-000096-80, EU CT Number 2024-517606-29-00
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.