
Abstract
Background:
Nearly half of patients with acute ischemic stroke who undergo intravenous thrombolysis (IVT) fail to achieve excellent functional outcomes. Early administration of tirofiban after IVT may improve patient outcomes.
Objective:
To evaluate the efficacy and safety of early tirofiban administration after intravenous tenecteplase in patients with acute ischemic stroke.
Methods and design:
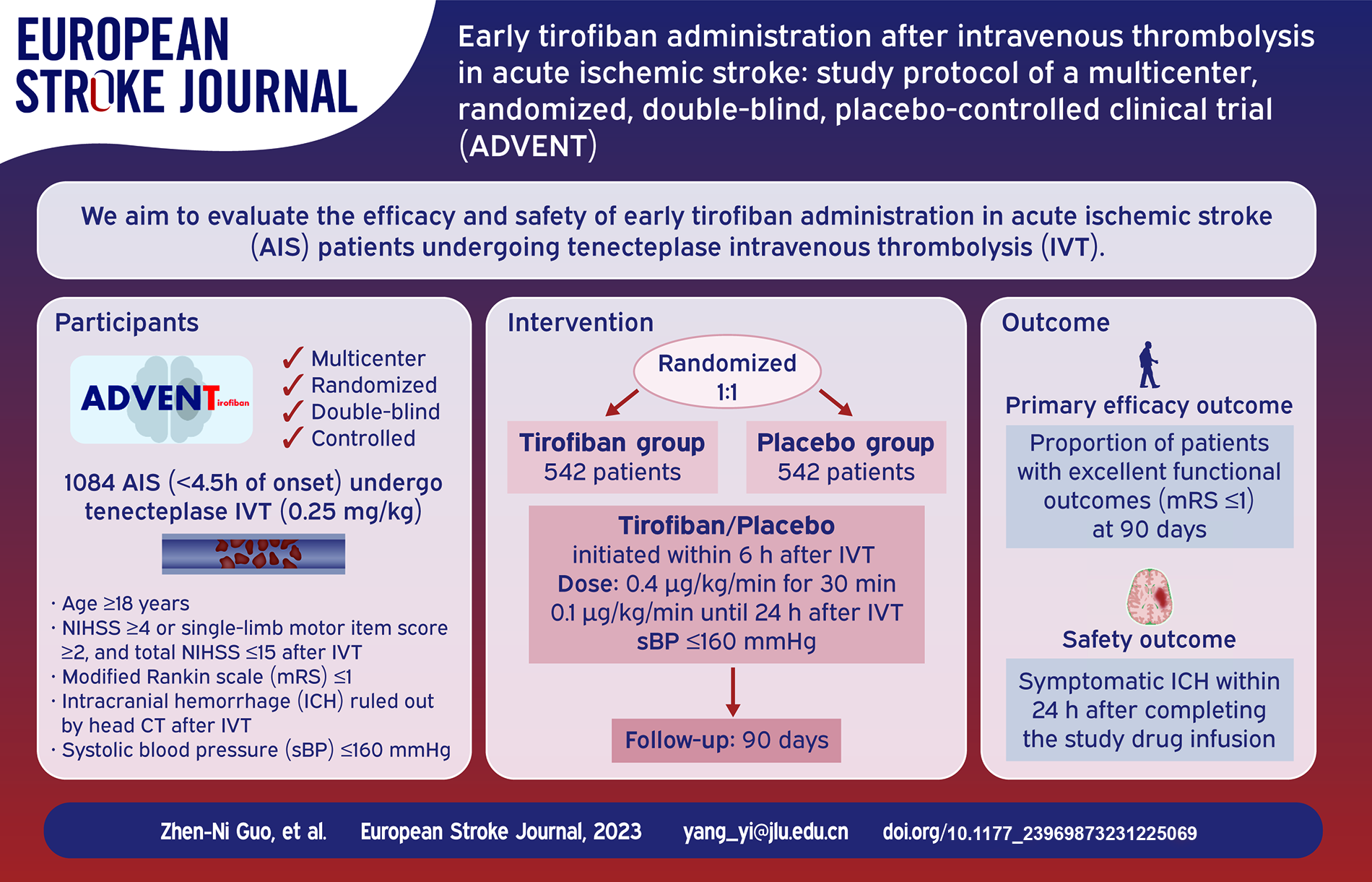
The ADVENT trial is a multicenter, randomized, parallel-controlled, double-blind clinical trial. A total of 1084 patients undergoing IVT without subsequent endovascular treatment will be recruited from multiple hospitals in China. Subjects will be randomized in a 1:1 ratio to receive tirofiban or placebo, which will be infused within 6 h after IVT until 24 h after IVT, at 0.4 μg/kg/min for 30 min and then at 0.1 μg/kg/min. The primary efficacy outcome is the proportion of patients with excellent functional outcomes (modified Rankin Scale (mRS) ⩽ 1) at 90 days. Secondary outcomes include the proportion of patients with favorable functional outcomes (mRS ⩽ 2) at 90 days and neurological functional assessments evaluated during hospitalization. Symptomatic intracranial hemorrhage will be the primary safety outcome. Mortality and other adverse events will be recorded.
Discussion:
This pivotal trial will provide important data on the early administration of antiplatelet therapy after IVT and may promote progress in treatment standards.
Trial registry:
ClinicalTrials.gov (NCT06045156)
Introduction and rationale
Currently, national guidelines recommend that antiplatelet drugs be administered 24 h after intravenous thrombolysis (IVT) to avoid the occurrence of symptomatic intracranial hemorrhage.1,2 However, delayed application of antiplatelet drugs can reduce the effectiveness of treatment, especially in patients who remain disabled after IVT. Therefore, more trials are needed to provide evidence of the safety and efficacy of antiplatelet agents administered within 24 h after IVT.
Tirofiban is a selective glycoprotein IIb/IIIa receptor inhibitor that reversibly inhibits platelet aggregation and thrombus formation. 3 It has been used in patients with myocardial infarction and acute ischemic stroke. 4 Since tirofiban takes effect quickly after treatment and the antiplatelet effect disappears quickly after drug withdrawal, it may be appropriate for application in patients after IVT.
Previous exploratory studies have reported that tirofiban may greatly improve the outcomes of patients who receive IVT.5–8 However, these studies were of small sample size, and the thrombolytic agent used was alteplase. Recently, several clinical trials reported that tenecteplase is non-inferior to alteplase in IVT of acute ischemic stroke, and because of its convenient administration, tenecteplase is promising as the first-choice thrombolytic agent.9–12 Therefore, the treatment mode of tenecteplase combined with early application of tirofiban warrants further investigation.
Thus, we designed the “Early tirofiban ADministration after intraVENous Thrombolysis in Acute Ischemic Stroke” (ADVENT) trial to evaluate the efficacy and safety of early tirofiban administration in patients receiving intravenous tenecteplase.
Methods
Design
The ADVENT trial is a prospective, multicenter, randomized, parallel-controlled, double-blind clinical trial. It was designed in compliance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines, approved by the Ethics Committee of the First Hospital of Jilin University, and registered at ClinicalTrials.gov (NCT06045156). The subjects will be recruited from multiple stroke centers in China.
Patient population
Inclusion criteria:
Age ⩾18 years old;
Clinically diagnosed with acute ischemic stroke and received standard dose (0.25 mg/kg) of tenecteplase IVT within 4.5 h of symptom onset;
Total National Institutes of Health Stroke Scale (NIHSS) ⩾4 or single-limb motor item score ⩾2, and total NIHSS ⩽15 after IVT;
Tirofiban or placebo can be initiated within 6 h after IVT administration;
Modified Rankin scale (mRS) score before onset ⩽1;
Intracranial hemorrhage ruled out by head computed tomography after IVT;
Systolic blood pressure before enrollment ⩽160 mmHg;
Informed consent obtained from patients or their legal representatives.
Exclusion criteria:
Received or about to undergo bridging therapy (subsequent endovascular treatment);
Large area of infarct as indicated by radiological imaging (at least one-third of middle cerebral artery territory);
Atrial fibrillation or suspected cardiac embolism;
With epileptic seizures;
Using antiplatelet, anticoagulant, or fibrinolytic agents within 24 h before recruitment;
Active bleeding or tendency to bleed after IVT;
Digestive system bleeding, urinary system bleeding, hemorrhagic retinopathy, or other systemic bleeding events within 1 year;
Severe renal or liver insufficiency; alanine aminotransferase or aspartate aminotransferase >3 times the upper limit of normal; creatinine clearance rate <30 mL/min, creatinine >200 μmol/L;
Life expectancy less than 3 months;
Pregnancy or lactation;
Known allergy to tirofiban;
Being enrolled or having been enrolled in other clinical trial within 3 months prior to this clinical trial;
Unwilling to be followed up or likely to have poor treatment compliance;
Other situations that the researcher deems unsuitable for inclusion in the study.
Randomization
Randomization will be performed via a web-based central randomization system, and patients will be stratified by centers into blocks of varying sizes. Eligible participants will be randomized 1:1 to the tirofiban or placebo group. All tirofiban and placebo will be packed in the same style, including labeling, dosage form, size, and color, except for a unique serial number. Each subject will be assigned a random serial number, and the corresponding medications will be used. The group assignment will be completely blinded to both the investigators and subjects.
Treatments or intervention
Tirofiban will be infused within 6 h after IVT at 0.4 μg/kg/min for 30 min and then at 0.1 μg/kg/min until 24 h after IVT. Normal saline will be administered in the placebo group in the same fashion. Systolic blood pressure will be maintained below or equal to 160 mmHg during the entire procedure in both groups, and antihypertensive agents can be used. Subjects will be treated according to current stroke management guidelines1,13 after completing the study drug infusion. Any other antiplatelet, anticoagulant, or fibrinolytic agent or medication with similar pharmacology will be prohibited within 24 h of IVT. A study flowchart is shown in Figure 1.
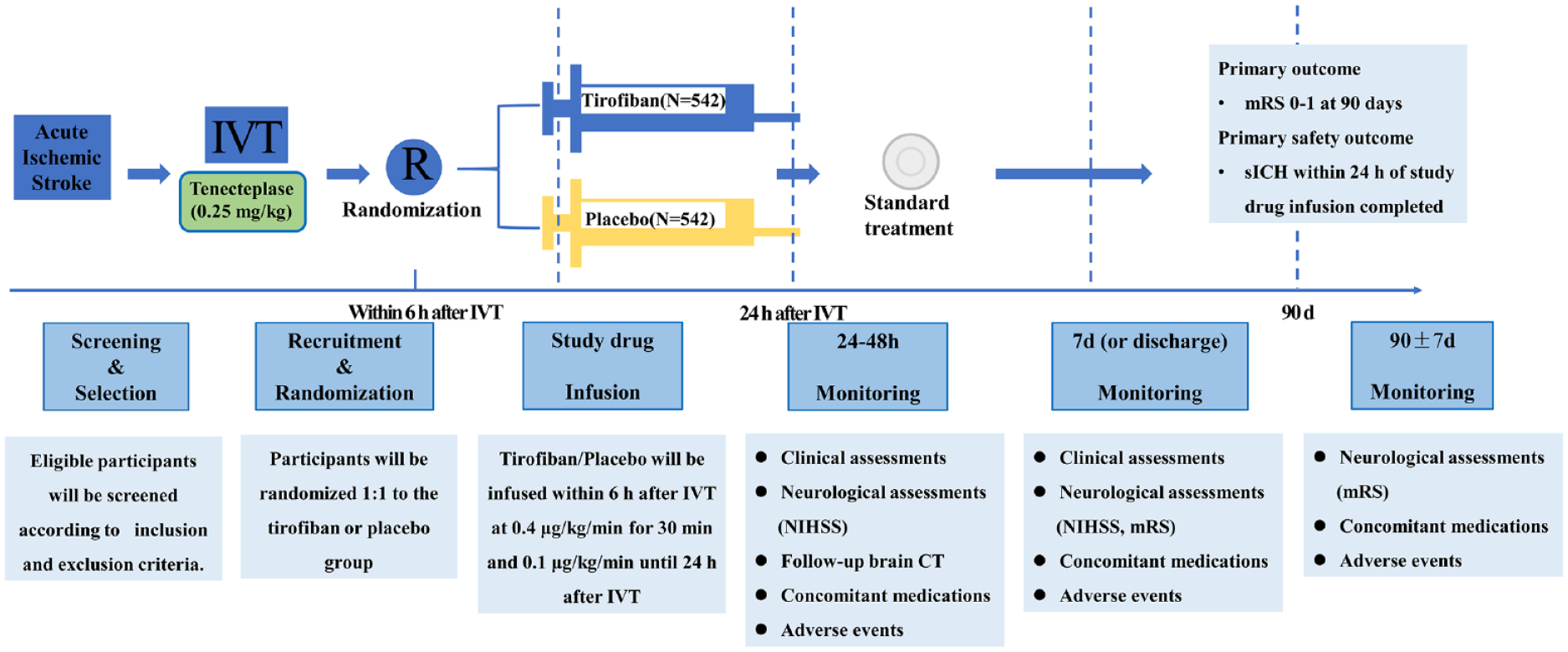
Study flowchart.
Follow-up time
Follow-up will be performed at 24–48 h, 7 days (or discharge if earlier), and 90 ± 7 days after IVT. Local clinical observers from stroke centers will be responsible for face-to-face visits with patients during hospitalization. Professional third-party observers will be responsible for follow-up at 90 ± 7 days. All observers will be blinded to treatment and group assignment.
Study outcomes
The primary efficacy outcome is the proportion of patients with excellent functional outcomes (mRS ⩽ 1) at 90 days. The secondary efficacy outcomes include the proportion of patients with favorable functional outcomes (mRS ⩽ 2), distribution of mRS at 90 days, and neurological function (assessed by NIHSS) at 24 h and 7 days (or discharge, if earlier) after IVT.
The primary safety outcome will be symptomatic intracranial hemorrhage (sICH) within 24 h after completing the study drug infusion. sICH is defined according to the European Cooperative Acute Stroke Study III classification as any hemorrhage with deterioration of 4 points or greater from the baseline NIHSS score. 14 Hemorrhagic transformation will also be recorded. Safety outcomes will include mortality and adverse events during the course of this study.
Data monitoring and safety committee
A data monitoring and safety committee (DMSC), consisting of neurologists and statisticians, has been established to evaluate the safety and credibility of the trial. The DMSC will meet annually and monitor the trial progress. In addition, to ensure the safety of the trial, safety outcomes and adverse events will be evaluated in the first 50 and 100 subjects. If there is a 10% or greater rate of sICH or mortality in a study arm, the study will be paused.
Sample size estimates
Interim analysis will be performed after 50% of the follow-up period is completed. The proportion of excellent functional outcomes in the control group in this trial was derived from the TRACE-2 trial, which reported that 62% of patients treated with tenecteplase had an mRS 0–1 at 90 days. 9 Previous studies have reported a 15% increase in mRS 0–1 with tirofiban administered after alteplase. 4 We hypothesized that the treatment effect might only reach two-thirds in the present study; hence, a 10% increase of mRS 0–1 was used to calculate the sample size. Considering a significance level of 0.05 and power of 90%, the sample size per group was estimated to be 487. We assume that the overall withdrawal or loss to follow-up rate will be 10%; therefore, 1084 participants (542 per group) will be required for this study. This estimation was performed using PASS software (NCSS, LLC. Kaysville, Utah, USA).
Statistical analysis
Student’s t-test or the Mann–Whitney U test will be used to compare continuous baseline variables, while the χ2 test or Fisher’s exact test will be used for categorical baseline variables. The outcomes will be estimated using a generalized linear mixed effect model with treatment as fixed effect, age, baseline NIHSS score, time from the end of thrombolysis to the administration of tirofiban and stroke etiology, defined by Trial of Org 10172 in Acute Stroke Treatment (TOAST) subtypes, as covariates, and the center as a random effect. Subgroup analyses will also be performed based on the prognostic factors or other variables of interest, including age (<65 years vs ⩾65 years), sex (Male vs female), baseline NIHSS score (<median vs ⩾median), time from the end of thrombolysis to the administration of tirofiban (<median vs ⩾median), and stroke etiology (TOAST subtypes). 15 All analyses will be performed using SPSS software (IBM Corp., Armonk, NY, USA) or R (R Core Team, 2014). Statistical significance was two-tailed and set at p < 0.05.
Discussion
Antiplatelet therapy is initiated 24 h after IVT and is recommended by many guidelines1,2; however, early application of antiplatelet drugs may be necessary in clinical practice. Whether and when to initiate antiplatelet therapy after IVT has long been discussed, but no consensus has been reached.1,13 The ADVENT trial is designed to provide more precise evidence in the early administration of antiplatelet therapy after IVT.
Some studies reported that patients who received tirofiban within 24 h after IVT showed better neurological function and better long-term outcomes without an increased risk of hemorrhage.5–8 For example, a study with 41 acute ischemic patients who were treated with tirofiban after alteplase IVT showed that these patients had better 3-month functional outcomes (mRS 0–1 70.7% vs 46.2%) and lower NIHSS scores at 7-day discharge (median 1 vs 6) compared to patients not receiving tirofiban after IVT, and did not increase the bleeding risk. 5 A study reported similar results of better functional outcomes and lower NIHSS scores in patients who were treated with tirofiban and reported that patients with the best outcome with tirofiban had initiation of the drug 2–12 h after IVT. 6 These studies form the basis for the design of the ADVENT trial. Furthermore, another study enrolled 948 patients and reported that tirofiban did not improve the outcome of patients who received endovascular treatment. 16 Thus, patients with subsequent endovascular treatment after IVT will be excluded in the present trial.
Our protocol has the following three advantages. First, we will select patients with NIHSS ⩾4 or single-limb motor item score ⩾2 after IVT. These patients are likely to have residual disability. Therefore, early antiplatelet therapy may enable them to recover to the greatest extent possible. Second, we will control the systolic blood pressure at ⩽160 mmHg throughout tirofiban administration to reduce the occurrence of hemorrhagic transformation. Third, we will select patients receiving tenecteplase for early tirofiban therapy. The short duration of tenecteplase administration will enable patients to receive tirofiban much earlier.
Summary and conclusions
In conclusion, we believe that the ADVENT trial is valuable and feasible. It will contribute greatly to the early administration of antiplatelet therapy after IVT and might promote progress in treatment standards.
Supplemental Material
sj-docx-1-eso-10.1177_23969873231225069 – Supplemental material for Early tirofiban administration after intravenous thrombolysis in acute ischemic stroke (ADVENT): Study protocol of a multicenter, randomized, double-blind, placebo-controlled clinical trial
Supplemental material, sj-docx-1-eso-10.1177_23969873231225069 for Early tirofiban administration after intravenous thrombolysis in acute ischemic stroke (ADVENT): Study protocol of a multicenter, randomized, double-blind, placebo-controlled clinical trial by Zhen-Ni Guo, Ke-Jia Zhang, Peng Zhang, Yang Qu, Reziya Abuduxukuer, Thanh N Nguyen, Hui-Sheng Chen and Yi Yang in European Stroke Journal
Footnotes
Acknowledgements
We would like to thank the Department of Clinical Research, the First Hospital of Jilin University.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by the National Natural Science Foundation of China (Grant No. 81971105), the Science and Technology Department of Jilin Province (YDZJ202201ZYTS677) to ZNG, and the Norman Bethune Health Science Center of Jilin University (2022JBGS03), Science and Technology Department of Jilin Province (YDZJ202302CXJD061, 20220303002SF), and Jilin Provincial Key Laboratory (YDZJ202302CXJD017) to YY.
Informed consent
The authors declare that they consent for publication.
Ethical approval
The Ethics Committee of the First Hospital of Jilin University approved this study.
Guarantor
YY.
Contributorship
YY and HSC researched literature and conceived the study. ZNG, KJZ, YQ, RA, and PZ were involved in protocol development, gaining ethical approval, patient recruitment and data analysis. ZNG and KJZ wrote the first draft of the manuscript. All authors reviewed and edited the manuscript and approved the final version of the manuscript.
Trial registration
ClinicalTrials.gov identifier: NCT06045156.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.