
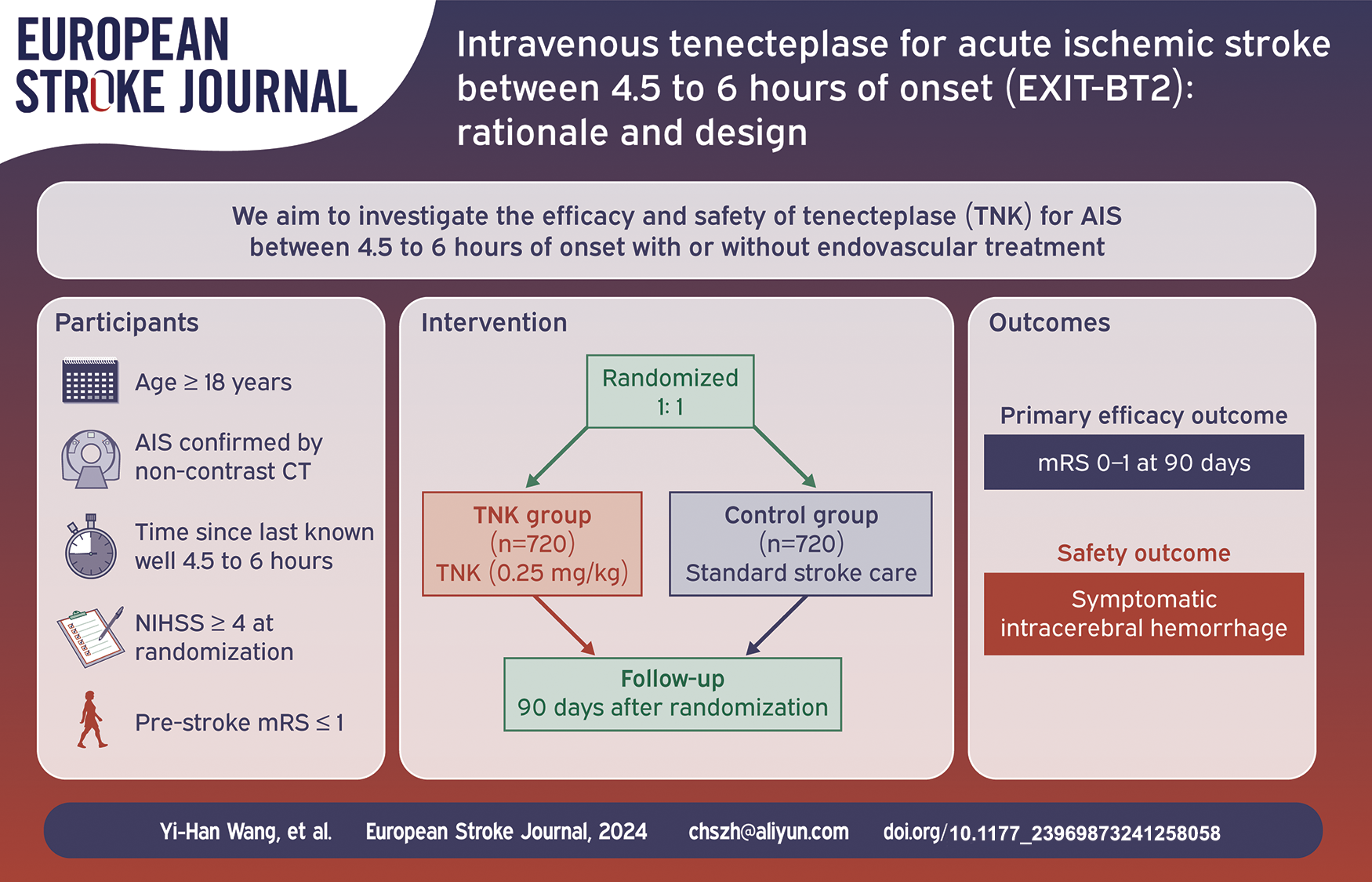
Abstract
Rationale:
To date, the benefit of intravenous thrombolysis for acute ischemic stroke (AIS) patients without advanced neuroimaging selection is confined to within 4.5 h of onset. Our phase II EXIT-BT (Extending the tIme window of Thrombolysis by ButylphThalide up to 6 h after onset) trial suggested the safety, feasibility, and potential benefit of intravenous tenecteplase (TNK) in AIS between 4.5 and 6 h of onset. The EXIT-BT2 trial is a pivotal study undertaken to confirm or refute this signal.
Aim:
To investigate the efficacy and safety of TNK for AIS between 4.5 and 6 h of onset with or without endovascular treatment.
Sample size estimates:
A maximum of 1440 patients are required to test the superiority hypothesis with 80% power according to a two-sided 0.05 level of significance, stratified by age, sex, history of diabetes, location of vessel occlusion, baseline National Institute of Health stroke scale score, stroke etiology, and plan for endovascular treatment.
Design:
EXIT-BT2 is a prospective, randomized, open-label, blinded assessment of endpoint (PROBE), and multi-center study. Eligible AIS patients between 4.5 and 6 h of onset are randomly assigned 1:1 into a TNK group or control group. The TNK group will receive TNK (0.25 mg/kg, a single bolus over 5–10 s, maximum 25 mg). The control group will receive standard medical care in compliance with national guidelines for acute ischemic stroke. Both groups will receive standard stroke care from randomization to 90 days after stroke onset according to national guidelines.
Outcome:
The primary efficacy endpoint is excellent functional outcome, defined as a modified Rankin Scale score 0–1 at 90 days after randomization, while the primary safety endpoint is symptomatic intracerebral hemorrhage, defined as National Institutes of Health Stroke Scale score increase ⩾4 caused by intracranial hemorrhage within 24 (−6/+12) h after randomization.
Conclusions:
The results of EXIT-BT2 may determine whether intravenous TNK has a favorable risk/benefit profile in AIS between 4.5 and 6 h of onset.
Introduction and rationale
Intravenous alteplase is strongly recommended to treat acute ischemic stroke (AIS) within 4.5 h of onset by current guidelines.1–3 Beyond 4.5 h of onset, advanced neuroimaging has been shown beneficial to guide intravenous thrombolysis use for selecting AIS patients.4–6 Considering its advantages as the latest generation of thrombolytic agents,7,8 tenecteplase (TNK) was widely investigated for treating AIS. Growing evidence shows that TNK is non-inferior to alteplase in the treatment of AIS within 4.5 h after symptom onset,9,10 is associated with higher recanalization rates and better functional outcomes as bridging therapy in mechanical thrombectomy patients with large vessel occlusion.11,12
Based on a multi-center trial, 13 urokinase has been considered safe for patients not eligible for alteplase to treat AIS between 4.5 and 6 h of onset by current Chinese Stroke guidelines (Class IIB). 3 In addition, the Third International Stroke Trial (IST-3) showed the beneficial trend of intravenous thrombolysis with alteplase within 6 h of onset, 14 but the benefit between 4.5 and 6 h of onset was not confirmed by an individual patient-level meta-analysis including 6 randomized controlled trials. 15 Our recent pilot phase II EXIT-BT (EXtending the tIme window of Thrombolysis by ButyphThalide up to 6 h after onset) trial investigated the effect of intravenous TNK for AIS between 4.5 and 6 h of onset and the results suggested the safety, feasibility and potential improvement of neurologic outcome. 16
In this context, we designed this phase III, randomized, open-label, blinded endpoint assessment, multi-center study to investigate the efficacy and safety of TNK for AIS between 4.5 and 6 h of onset.
Methods
Design
EXIT-BT2 is a prospective, randomized, open-label, blinded endpoint assessment, multicenter study in China assessing the efficacy and safety of TNK for AIS between 4.5 and 6 h of onset.
Study population
In the EXIT-BT2 trial, eligible participants are AIS patients who are treated between 4.5 and 6 h of symptom onset. They will be enrolled at approximately 50 sites in China between October 2023 and June 2025. The detailed inclusion/exclusion criteria are listed in Table 1.
Study entry criteria.

mRS: modified Rankin Scale; NIHSS: National Institutes of Health Stroke Scale; INR: international normalized ratio; APTT: activated partial thromboplastin time.
Standard protocol approvals, registration, and patient consent
The EXIT-BT2 trial is registered on www.clinicaltrial.gov (NCT06010628). The protocol and data collection of the trial have or will have been approved by the ethics committee of the General Hospital of Northern Theater Command and all participating sites. All patients or their representatives will provide written informed consent before inclusion into the trial.
Randomization and intervention
Eligible patients will be assigned into the TNK group or control group in a 1:1 ratio using central and computerized random sequence generation stratified by center (Figure 1). The TNK group will receive TNK (0.25 mg/kg, a single bolus over 5–10 s, a maximum of 25 mg). The control group will receive standard medical care in compliance with national guidelines for acute ischemic stroke. Endovascular treatment (EVT) is allowed for both groups if the stroke is due to large vessel occlusion. Both groups will receive standard stroke care from randomization to 90 days after stroke onset according to national guidelines. 3
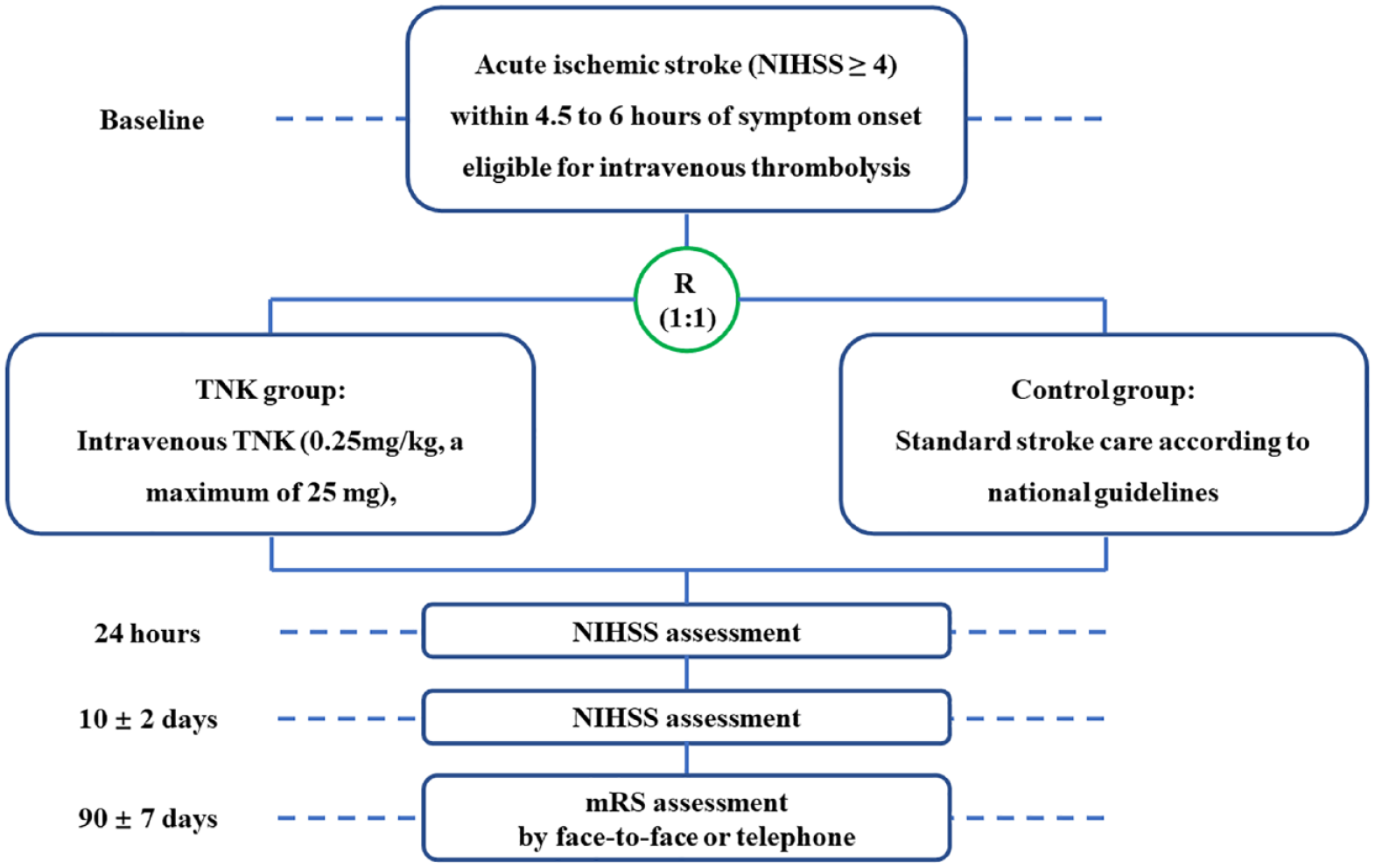
Study flowchart.
Outcomes
The primary endpoint is excellent outcome, defined as a modified Rankin Scale (mRS) score 0–1 at 90 days.
There are seven secondary efficacy endpoints that will be analyzed in a stepwise gatekeeper manner to control for multiplicity. 17 The hierarchical order of analysis will be: (1) all-cause mortality within 90 ± 7 days; (2) functional independence (mRS score 0–2) at 90 days; (3) level of disability (ordinal distribution of mRS) at 90 days; (4) change in National Institute of Health stroke scale (NIHSS) score compared with baseline at 24 (−6/+12) h; (5) change in NIHSS score compared with baseline at 10 ± 2 days; (6) early neurological improvement (ENI), defined as >4-point decrease in NIHSS within 24 (−6/+12) h; (7) new stroke or other vascular event(s) at 90 ± 7 days.
The safety endpoints include (1) symptomatic intracranial hemorrhage within 24 (−6/+12) h, defined as parenchymal hematoma type 1 or 2 (PH1 or PH2), remote intraparenchymal hematoma (RIH), subarachnoid hemorrhage, or intraventricular hemorrhage on head CT/MRI scan causally associated with clinically significant neurological deterioration (NIHSS score ⩾ 4 point increase) in the opinion of the clinical investigator or independent safety monitor 18 ; (2) any radiologic intracranial hemorrhage within 24 (−6/+12) h according to the Heidelberg Bleeding Classification 19 ; (3) major systemic bleeding event within 24 (−6/+12) h, defined as fatal bleeding, and/or symptomatic bleeding in a critical area or organ, such as intraspinal, intraocular, retroperitoneal, intra-articular or pericardial, or intramuscular with compartment syndrome, and/or bleeding causing a fall in hemoglobin levels of 1.24 mmol/L or more, or leading to a transfusion of 2 U or more of whole blood or red cells 20 ; (4) any systemic bleeding event within 24 (−6/+12) h.
Follow-up procedure
Study visits will be performed at 24 h, 10 and 90 days after randomization (Table 2). At baseline, demographic characteristics, routine laboratory tests, and neuroimaging will be collected and NIHSS and prestroke mRS will be evaluated. At 24 h, a repeat non-contrast CT/MRI will be performed in the TNK group. NIHSS scoring will be repeated at 24 h and 10 days or at discharge (whichever comes earlier), and mRS will be assessed at 10 days (or at discharge if earlier) and 90 days. All clinical assessments including NIHSS and mRS will be evaluated by certified assessors according to a standardized procedure manual in each study center. Baseline and follow-up NIHSS scores will be evaluated by the same neurologist who is not blinded to treatment allocation. mRS will be mainly evaluated in person or by telephone interview (if a face-to-face interview is not possible) by one trained and certified staff in each center who is unaware of the randomized treatment assignment. All mRS assessment will be recorded on video for central adjudication. Concomitant medications and adverse events within 90 days after randomization will be recorded in detail by investigators and adjudicated by certified assessors. The cycle of this research is expected to be about 24 months.
Timing of all assessment procedures in EXIT-BT2.
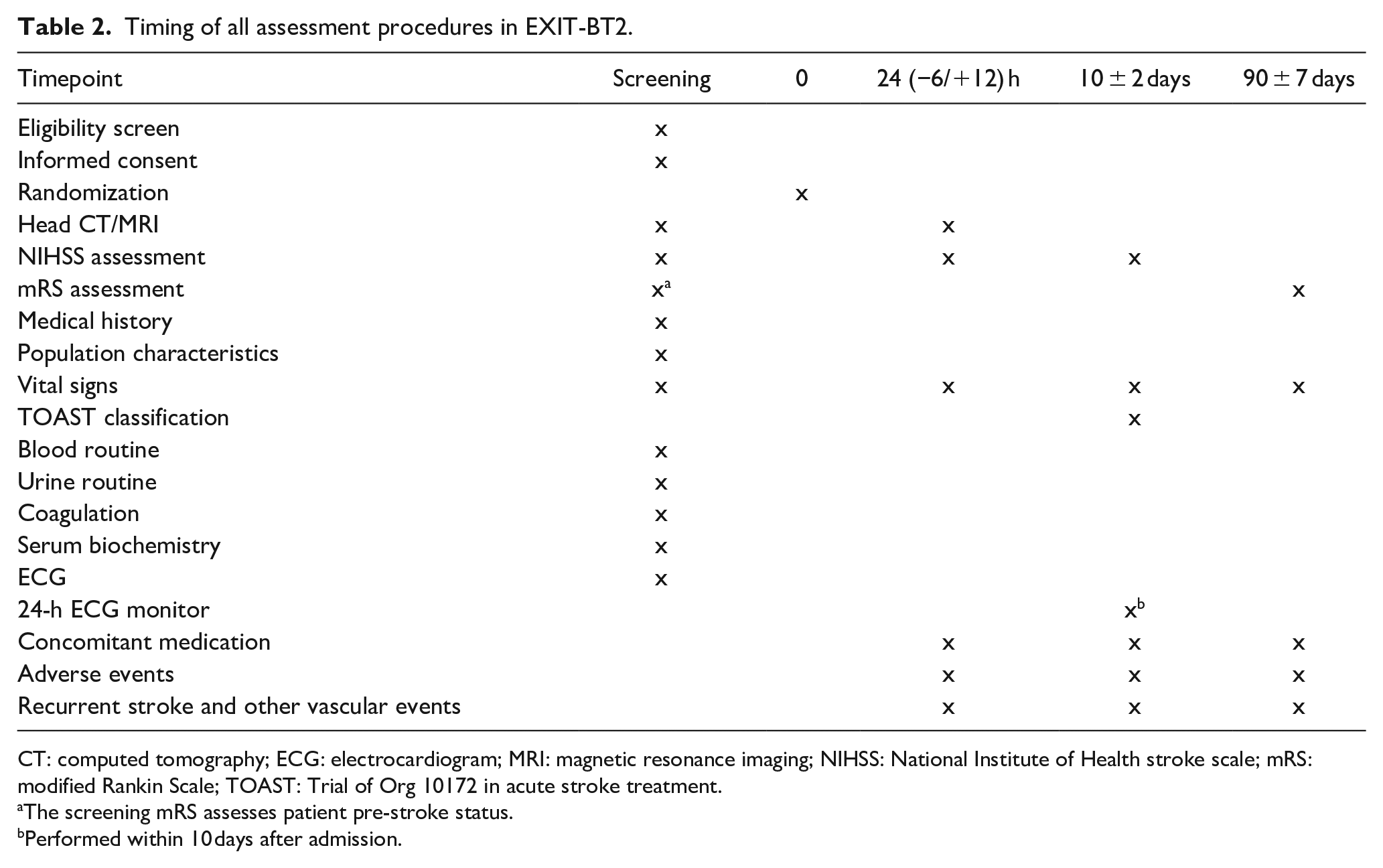
CT: computed tomography; ECG: electrocardiogram; MRI: magnetic resonance imaging; NIHSS: National Institute of Health stroke scale; mRS: modified Rankin Scale; TOAST: Trial of Org 10172 in acute stroke treatment.
The screening mRS assesses patient pre-stroke status.
Performed within 10 days after admission.
Quality control
The Data Safety and Monitoring Committee (DSMC) will perform data verification every 6 months in this study.
Data management and monitoring
All data are recorded at electronic data capture (EDC) system (http://EXIT-BT2.medsci.cn) that will store the patient case report form. The enrolled patients will be randomly assigned into the experimental group and control group at a ratio of 1:1. The data will be downloaded from EDC with a dedicated statistician for statistical analysis. An independent DSMC is established to ensure ongoing monitoring of patient safety, such as hemorrhagic events, and other adverse events (AEs). An interim analysis will be performed after 50% of patients have been followed up completely by an independent statistician of the DSMC, who is not involved in managing the trial. The stopping rule or sample size adjusting rule will be prespecified by the investigators and the main study statistician. Based on the analysis, the DSMC can recommend to the Steering Committee of the EXIT-BT2 trial to adjust the sample size, remove a subgroup of patients, or terminate the study early. A recommendation of early termination due to safety is that the proportion of sICH is more than 5% higher in the TNK group than in the control group. A statistical stopping guideline for the primary efficacy endpoint is an overwhelming benefit (alpha-level of 0.0132) or the treatment effect of less than 3.5% (absolute difference of TNK vs control).
Adverse event monitoring
An AE is any untoward medical occurrence that occurs during the study. All information about AEs will be recorded on the AE page of the case report form including site investigator ratings of severity and expectedness. Whether unexpected AEs are potentially causally associated with the active group study treatment will be further adjudicated by the overall trial principal investigator.
Sample size determination
For sample size calculations, the proportion of expected excellent functional outcome (mRS 0–1) at 90 days in the control group is estimated to be 69.0%, based on the EXIT-BT pilot study. 16 An absolute increase of 7.0% in this proportion is projected for the study intervention, yielding an expected experimental group mRS 0–1 rate of 76.0%. Using a power = 80% and α = 0.05 to carry out the two-side test, the calculated sample size to test the superiority hypothesis is 1270. In consideration of 10% lost to follow-up and an alpha consumption by an interim analysis (O’Brien-Fleming method), the total sample size is therefore readjusted to 1440. Therefore, this study will include 1440 patients, with 720 patients in each group.
Statistical analysis
An intention-to-treat (ITT) analysis will be used to analyze outcome differences between the two groups. A per protocol sensitivity analysis will also be performed. All data will be analyzed with SPSS 23.0 Software. The mean ± standard deviation (SD) will be used if the data are normally distributed, and the median and interquartile range (IQR) will be used if the data are non-normally distributed. Count data will be expressed as n (%). Binary outcomes, including the primary mRS 0–1 at 90 days endpoint and secondary endpoints such as mRS (0–2) at 90 days, incidence of ENI, proportion of sICH within 24 h, incidence of any intracranial hemorrhage within 24 h, and occurrence of all-cause death in 90 days, will be compared in the active versus control groups using binary logistic regression. Distribution of the mRS at 90 days between the two groups will be compared using ordinal logistic regression. Change in NIHSS score between the two groups will be compared using the general linear model. Time-to-event of stroke recurrence and other vascular events will be compared using Cox regression. There is statistical significance if a two-sided p-value <0.05. The seven secondary efficacy endpoints will be analyzed in a stepwise gatekeeper manner to control for multiplicity.
The lead analyses of the primary and secondary efficacy endpoints in EXIT-BT2 will stratified by age (<60 years vs ⩾60 years), sex (female vs male), history of diabetes (yes vs no), location of index vessel occlusion (anterior vs posterior circulation), NIHSS score at randomization (4–10 vs ⩾11), stroke etiology (large-artery atherosclerosis, cardioembolic, small-artery occlusion, other determined cause, and undetermined cause), EVT (yes vs no), and plan at time of randomization to pursue endovascular treatment (yes vs no). Unadjusted analyses will be performed as sensitivity analyses.
Analysis for heterogeneity of treatment effect upon the primary endpoint will be conducted in seven stratification subgroups. Differences in the primary endpoint across each subgroup will be assessed by testing for interaction of the preset baseline variable with the primary endpoint.
Study organization and funding
The protocol was designed by Hui-Sheng Chen and discussed by the Trial Steering Committee. The Trial Steering Committee includes external scientific advisors, and will monitor the research and data regularly. The Trial Steering Committee will organize teleconference or physical meetings to give recommendations about the trial. Neuroimaging associated with clinical events will be collected centrally and interpreted by two independent neuroradiologists. The trial is initiated and supported by Cerebrovascular Disease Collaboration & Innovation Alliance (CDCIA) of Liaoning. TNK will be donated by CSPC Recomgen Pharmaceutical Co., LTD.
Current status
The trial began enrollment on February 1, 2024. As of the time of this submission, 10 centers were activated, and 13 patients were enrolled. Recruitment will be continued until the complete sample size is achieved.
Discussion
As an effective treatment for acute ischemic stroke, IVT is strongly recommended by guidelines, but the time window is confined to 4.5 h of last known well without advanced imaging selection. The EXIT-BT phase II trial studied the effect of intravenous TNK in AIS between 4.5 and 6 h of last known well. The results suggested good safety and a promising benefit of TNK in this population.
In the EXIT-BT trial, 16 the 2.0% rate of sICH in the TNK group was comparable to those reported in previous studies (3% in IST3 14 ; 2–3% in NORTEST 21 ; 2.4% in WAKE-UP 5 ; 2% in TREAC-II 10 ). With regard to efficacy, the 8.2% difference in excellent functional outcome was comparable to that in the ECASS III (7.2%), 18 the WAKE-UP (11.5%) 5 and the TWIST trial (7.0%), 22 but higher than the treatment difference observed in the 4.5–6 h subgroup in the IST3 (4.7%), 14 EXTEND trial (5.9%) 4 and meta-analysis including alteplase thrombolysis (2.0%). 12 The small difference in the meta-analysis may be attributed to the more severe neurological deficit in these included studies (median NIHSS of 11–13), given that severe neurological deficit is likely due to large vessel occlusion, which may result in a lower likelihood of benefit of alteplase in this population. The current EXIT-BT2 trial is expected to enroll patients with moderate neurological deficit, given the moderate neurological deficit in the pilot EXIT-BT trial (median NIHSS of 5), TRACE II (median NIHSS of 7), 10 and PROST trial (median NIHSS of 6) 23 in China. Nevertheless, the current sample size calculation based on this pilot EXIT-BT may still be insufficient, given the small treatment effect in the meta-analysis. 12
In EXIT-BT, Butyphthalide was used, which may affect the treatment effect of TNK although there was no evidence of an interaction between Butyphthalide and TNK. In agreement with excellent functional outcome favoring TNK, more ENI was found in the TNK group (20.4% vs 4.1%). 16 Altogether, EXIT-BT suggested that among AIS patients presenting between 4.5 and 6 h of onset, intravenous TNK seemed safe, feasible, and improved early neurological outcome, with the potential of improving 3-month neurological functional outcome, although recent TWIST trial data did not demonstrate the benefit of intravenous TNK in patients with wake-up stroke selected with non-contrast CT and within 4.5 h of awakening. 22
Considering the promising results of EXIT-BT as well as supporting evidence of previous studies,13,14 this phase III, large sample size, multicenter EXIT-BT2 trial is designed to confirm the benefit of intravenous TNK in AIS patients between 4.5 and 6 h of last known well and without advanced neuroimaging selection. This trial has several distinct characteristics from previous studies, including study of patients in the 4.5-to-6 h time window, no requirement for advanced neuroimaging selection, and the use of TNK.
Conclusion
The results of EXIT-BT2 may determine whether intravenous TNK has a favorable risk/benefit profile in acute ischemic stroke patients within 4.5–6 h of onset and without advanced neuroimaging selection.
Footnotes
Acknowledgements
We are grateful to the Trial Steering Committee and Data Safety and Monitoring Committee for their contributions to this study.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: There was no conflict of interest with CSPC Recomgen Pharmaceutical Co., LTD. T.Nguyen discloses advisory board for Aruna Bio, Brainomix; Associate Editor of Stroke.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by grants from the Science and Technology Project Plan of Liao Ning Province (2022JH2/101500020).
Ethics approval
This retrospective study was approved by the Institutional Review Board of General Hospital of Northern Theater Command (IRB: Y (2023) 144).
Informed consent
All patients or their representatives will provide written informed consent before inclusion into the trial.
Guarantor
I will take full responsibility for the article, including for the accuracy and appropriateness of the reference list. Submitting author (Hui-Sheng Chen).
Contributorship
Y.H.W., Z.N.G. M.R.C., and Z.G.Y. wrote the first draft of the manuscript. T.N.N. and J.L.S. critically revised the manuscript. Y.Y. and H.S.C. designed the study and critically revised the manuscript. All the authors have carefully read and approved the article.
Data availability statement
Not applicable.