
Abstract
Rationale
Cerebrovascular diseases associated with pregnancy and postpartum period are uncommon; however, they can have an important impact on health of both women and foetus or newborn.
Aims
To evaluate the frequency, characteristics and management of cerebrovascular events in pregnant/postpartum women, to clarify pathophysiological mechanisms underlying the occurrence of these events including biomolecular aspects, and to assess the short- and long-term cerebrovascular and global cardiovascular outcome of these patients, their predictors and infant outcome.
Methods and design
This is an observational, prospective, multicentre, international case–control study. The study will include patients with cerebrovascular events during pregnancy and/or within six months after delivery. For each included case, two controls will be prospectively recruited: one pregnant or puerperal subject without any history of cerebrovascular event and one non-pregnant or non-puerperal subject with a recent cerebrovascular event. All controls will be matched by age, ethnicity and type of cerebrovascular event with their assigned cases. The pregnant controls will be matched also by pregnancy weeks/trimester. Follow-up will last 24 months for the mother and 12 months for the infant.
Summary
To better understand causes and outcomes of uncommon conditions like pregnancy/postpartum-related cerebrovascular events, the development of multisite, multidisciplinary registry-based studies, such as the Stroke in Pregnancy and Postpartum study, is needed in order to collect an adequate number of patients, draw reliable conclusions and give definite recommendations on their management.
Introduction
In the last decades, stroke associated with pregnancy and postpartum period has been studied in relation to women’s health and the consequences for the foetus or newborn.1,2 The condition is not common; reported estimates of the incidence of strokes related to pregnancy vary between 4 and 40 per 100,000 deliveries and appears to be further increasing.3–8 This is likely to be explained by women delaying pregnancy until older ages and by increasing rates of obesity, diabetes and hypertension. 8 In addition, this condition is poorly characterised and the consequences are devastating. Pregnancy-associated stroke is the most common cause of serious long-term disability after pregnancy. 8 Therefore, more studies are needed. Stroke related to pregnancy includes ischemic and haemorrhagic cerebrovascular (CBV) events and cerebral venous thrombosis (CVT). Moreover, other CBV disorders such as posterior reversible encephalopathy syndrome (PRES) or reversible cerebral vasoconstriction syndrome (RCVS) may occur as related to pregnancy particularly in the peripartum or puerperium.9–14
Potential risk factors for pregnancy-related stroke include hypertensive disorders of pregnancy, such as preeclampsia and eclampsia, and a prothrombotic state, which increases the risk of arterial and venous thrombosis. 8 However, it is still controversial if pregnancy should be considered a risk factor for stroke, although for some CBV conditions such as CVT there is strong epidemiological evidence that puerperium represents a risk factor.15–19 Some of the CBV events may be coincidental while others may be triggered by pregnancy itself, but the data on this issue are still not clear. Additionally, few data are available regarding acute treatments, appropriate diagnostic tools and preventive treatment in pregnancy-related CBV events.
The Stroke in Pregnancy and Postpartum (SiPP) study will try to fill the knowledge gap in this field. It is supported by the ESO (European Stroke Organisation) 20 WISE (Women Initiative of Stroke in Europe) group that has the mission to promote international multicentre and multidisciplinary research collaborations (involving neurologists, neurosurgeons, neuroradiologists, epidemiologists, gynaecologists/obstetricians, paediatricians, anaesthesiologists and geneticists) in order to address unresolved questions/issues regarding stroke in women.
Methods
Objectives
The primary objective of the SiPP study is to describe characteristics and course of pregnancy-related CBV disorders: frequency, pathophysiological mechanisms, clinical and neuroimaging characteristics, associated risk factors, acute interventions, secondary prevention treatments, short-term outcome and foetal/newborn outcome. The secondary objectives are to evaluate long-term outcomes in women with pregnancy-related CBV disorders, to individuate potential predictors of outcome and to assess foetus/newborn/infant outcome. Furthermore, this study aims to increase awareness and knowledge of pregnancy-related CBV disorders and to establish a sample of data which can be used for other nested studies.
Finally, if the study will be awarded research funding, an additional objective would be to establish a bio-bank of biological samples from women with pregnancy/postpartum-related CBV events. Therefore, an ad hoc sub-study will be planned for performing specific molecular and genetic analysis, to better understand the pathophysiological processes underlying these disorders.
Design
This is an observational, prospective, multicentre, international case–control study (see the checklists in the supplemental material). Due to the relative rarity of the target condition, the registry will also give the opportunity to separately enter data collected retrospectively for those centres that have these data available. If the SiPP study will be awarded research funding, a sub-study collecting bio-bank molecular/genetics data will be conducted in patients who will give their informed consent. In these patients, specific blood samples will be drawn at baseline of the index CBV event.
Table 1 reports the study flow chart with the schedule of clinical assessments, and Table 2 reports the study variables to be collected at baseline and follow-up. Inclusion period will last 24 months. Follow-up after the CBV event will last 24 months for the mother and 12 months for the infant.
Flow chart of study procedures.
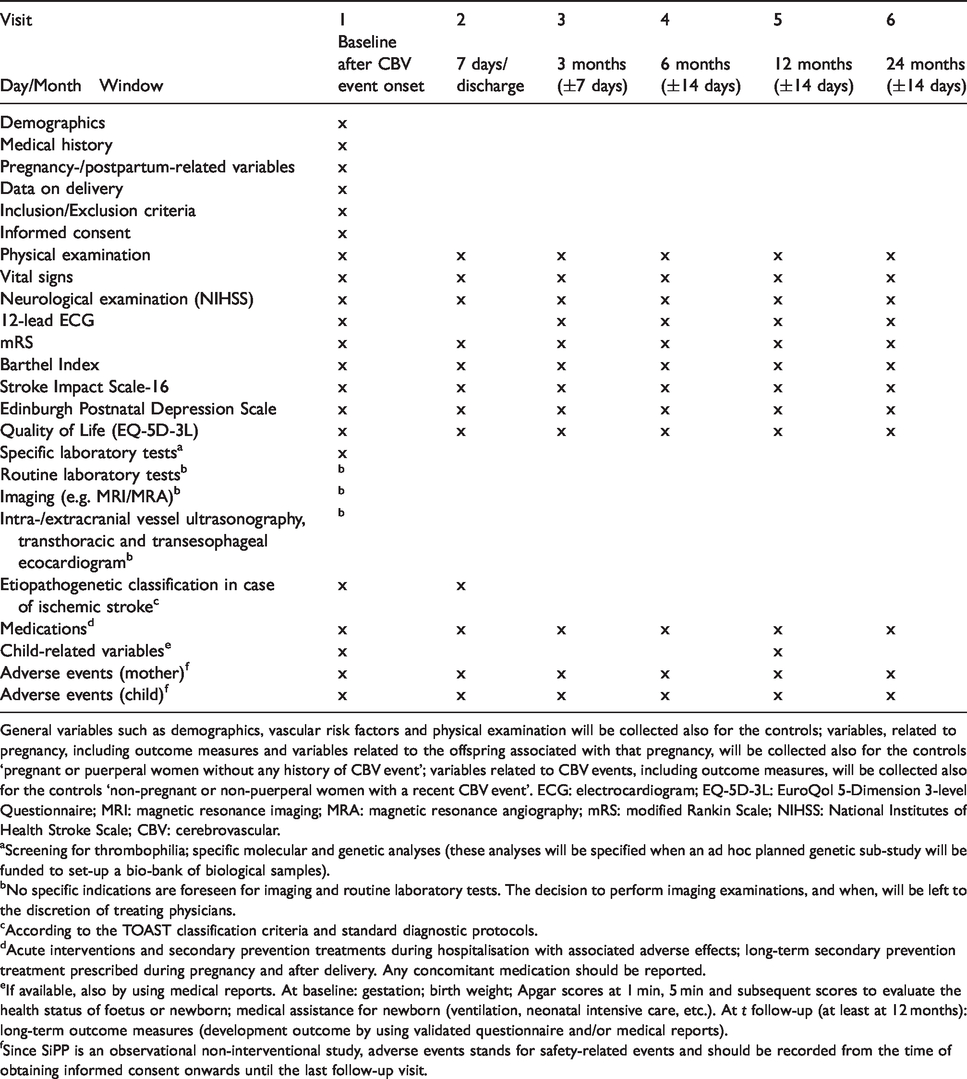
General variables such as demographics, vascular risk factors and physical examination will be collected also for the controls; variables, related to pregnancy, including outcome measures and variables related to the offspring associated with that pregnancy, will be collected also for the controls ‘pregnant or puerperal women without any history of CBV event’; variables related to CBV events, including outcome measures, will be collected also for the controls ‘non-pregnant or non-puerperal women with a recent CBV event’. ECG: electrocardiogram; EQ-5D-3L: EuroQol 5-Dimension 3-level Questionnaire; MRI: magnetic resonance imaging; MRA: magnetic resonance angiography; mRS: modified Rankin Scale; NIHSS: National Institutes of Health Stroke Scale; CBV: cerebrovascular.
aScreening for thrombophilia; specific molecular and genetic analyses (these analyses will be specified when an ad hoc planned genetic sub-study will be funded to set-up a bio-bank of biological samples).
bNo specific indications are foreseen for imaging and routine laboratory tests. The decision to perform imaging examinations, and when, will be left to the discretion of treating physicians.
cAccording to the TOAST classification criteria and standard diagnostic protocols.
dAcute interventions and secondary prevention treatments during hospitalisation with associated adverse effects; long-term secondary prevention treatment prescribed during pregnancy and after delivery. Any concomitant medication should be reported.
eIf available, also by using medical reports. At baseline: gestation; birth weight; Apgar scores at 1 min, 5 min and subsequent scores to evaluate the health status of foetus or newborn; medical assistance for newborn (ventilation, neonatal intensive care, etc.). At t follow-up (at least at 12 months): long-term outcome measures (development outcome by using validated questionnaire and/or medical reports).
fSince SiPP is an observational non-interventional study, adverse events stands for safety-related events and should be recorded from the time of obtaining informed consent onwards until the last follow-up visit.
Variables to be collected at baseline and follow-up. a

BI: Barthel Index; BMI: body mass index; BT: body temperature; CBV: cerebrovascular; CVT: cerebral venous thrombosis; DBP: diastolic blood pressure; ECG: electrocardiogram; EQ-5D-3L: EuroQol 5-Dimension 3-level Questionnaire; HR: heart rate; mRS: modified Rankin Scale; NIHSS: National Institutes of Health Stroke Scale; QoL: quality of life; SBP: systolic blood pressure; SIS-16: Stroke Impact Scale-16; CBV: cerebrovascular.
aGeneral variables such as demographics, vascular risk factors, physical examination, will be collected also for the controls; variables, related to pregnancy, including outcome measures and variables related to the offspring associated with that pregnancy, will be collected also for the controls ‘pregnant or puerperal women without any history of CBV event’; variables related to CBV events, including outcome measures, will be collected also for the controls ‘non-pregnant or non-puerperal women with a recent CBV event’.
bNo specific indications are foreseen for imaging. The decision to perform imaging examinations, and when, will be left to the discretion of treating physicians. In any case, it is supposed that at least a baseline neuroimaging is performed in order to make a specific diagnosis.
An ad hoc secure web-based database (eCRF, electronic case report form) will be established to allow collection of standardised data from centres of the different countries. All data will be anonymised for privacy reasons, and each patient included in the study will be identified by an ID number. Effort in collecting outcome data as much as possible will be made also by telephone interviews or medical records, if in-person follow-up visits will not be possible.
A centralised neuroimaging analysis will be planned on appropriately anonymised neuroimages. If clinically indicated, patients will undergo magnetic resonance imaging (MRI) at study inclusion at baseline including the following sequences: T1, T2, FLAIR, DWI, PWI, GRE or SWI, MR-angiography (venous and/or arterial phase). On the available MRIs, the presence of the following radiological signs, besides the index CBV event, will be assessed for each cases and control at study inclusion: white matter hyperintensity (by modified Fazekas Scale or semiautomated volumetric methods) 21 including silent lacunar infarcts; cerebral microbleeds (by Brain Observer MicroBleeds scale, BOMBS) 22 ; dilated perivascular spaces 23 ; superficial siderosis; blood–brain barrier alterations; cerebral oedema; haemorrhagic transformation (as per National Institute of Neurological Disorders and Stroke (NINDS), European Cooperative Acute Stroke Study (ECASS) and Safe Implementation of Thrombolysis in Stroke-Monitoring Study (SITS-MOST) criteria in case of ischemic stroke)24–26; diagnosis of cerebral amyloid angiopathy (Boston criteria) 27 and brain atrophy. 28
Study population
The study will include patients who have had CBV events during pregnancy and/or within six months after delivery in order to include the acute (i.e. the first 6–12 h after childbirth), subacute (starting after the acute postpartum period concludes, can last for 2–6 weeks) 29 and even the delayed (starting after the subacute postpartum period, lasts up to six months) postpartum period. The considered events will be ischemic stroke (including clinical transient ischemic attack (TIA) with evidence of cerebral infarction on neuroimaging corresponding to patient symptoms), haemorrhagic stroke (including primary and secondary (e.g. due to vascular malformations, systemic disorders) intracerebral haemorrhage (ICH), subarachnoid haemorrhage (SAH)), CVT, PRES or RCVS. For each included case, two controls will be prospectively recruited: one pregnant or puerperal woman without any history of CBV event and one non-pregnant or non-puerperal woman with a recent CBV event. Controls will be recruited in the same hospital admitting the index patient. All controls will be matched by age (±2 years), ethnicity and type of CBV event with their assigned cases. The pregnant controls will also be matched by pregnancy weeks/trimester. General variables such as demographics, vascular risk factors, physical examination will be collected also for the controls; variables, related to pregnancy, including outcome measures and variables related to the offspring associated with that pregnancy, will be collected also for the controls ‘pregnant or puerperal women without any history of CBV event’; variables related to CBV events, including outcome measures, will be collected also for the controls ‘non-pregnant or non-puerperal women with a recent CBV event’.
Patients with TIA without evidence of cerebral infarction on neuroimaging corresponding to patient symptoms and patients with traumatic ICH and/or traumatic SAH will be excluded.
Randomisation
Since SiPP is an observational study, no randomisation will be done.
Treatment or intervention
As SiPP is a non-interventional study, no specific treatment or intervention will be investigated. No changes to current management of included patients are required. The Steering Committee of the study will promote adherence to available guidelines and best medical practice.
Outcomes
Outcome measures are listed in Table 3. Both short- and long-term outcomes, including specific safety outcomes, will be evaluated for both the mother and the infant. In particular, short-term outcomes will be assessed during hospital admission (such as in-hospital mortality, functional outcome at discharge, length of hospital stay and discharge destination) and long-term outcomes for the mother (e.g. functional outcome, mortality, recurrence of CBV event, incidence of other cardiovascular events, post-partum depression, breastfeeding issues, subsequent pregnancies and quality of life (QoL)) at 3, 6, 12 and 24 months. For the infant, development outcome will be evaluated at least at 12 months. Functional outcome will be assessed using the modified Rankin Scale (mRS) score, Barthel Index (BI) and Stroke Impact Scale-16 (SIS-16) 30 by in-person visit or telephone interview, with favourable outcome defined as mRS = 0–2, BI ≥95 and SIS-16 ≥ 80. Postpartum depression and QoL will be assessed by using Edinburgh Postnatal Depression Scale and EuroQol 5-Dimension 3-level Questionnaire, respectively. Specific safety endpoints for the mother include vaginal bleeding occurring after reperfusion/recanalisation treatment in cases of acute ischemic stroke or after any other treatments, miscarriages during the index CBV event, deep venous thrombosis and pulmonary embolism. The incidence of haemorrhagic transformation of the index cerebral infarct due to arterial occlusion or CVT will be evaluated as well as the incidence of symptomatic intracranial haemorrhage defined primarily as any bleeding associated with any neurological deterioration measured with the National Institutes of Health Stroke Scale (NIHSS) as per the NINDS criteria. 24 ECASS (i.e. any haemorrhage plus a neurological deterioration of ≥4 points on the NIHSS from baseline, or from the lowest NIHSS value after baseline to seven days or leading to death) 25 and SITS-MOST (i.e. local or remote parenchymal haemorrhage type 2 on the 22 to 36 h post-treatment imaging scan, combined with a neurological deterioration of ≥4 points on the NIHSS score from baseline, or from the lowest NIHSS score between baseline and 24 h or leading to death) 26 criteria will also be used for defining symptomatic intracerebral haemorrhage.
Outcome measures. a

aOutcome measures related to pregnancy, including those related to the offspring associated with that pregnancy, will be collected also for the controls ‘pregnant or puerperal women without any history of CBV event’; outcome measures related to CBV events will be collected also for the controls ‘non-pregnant or non-puerperal women with a recent CBV event’. CBV: cerebrovascular; CVT: cerebral venous thrombosis; EQ-5D-3L: EuroQol 5-Dimension 3-level Questionnaire; mRS: modified Rankin Scale; QoL: quality of life; NINDS: National Institute of Neurological Disorders and Stroke; ECASS: European Cooperative Acute Stroke Study; SITS-MOST: Safe Implementation of Thrombolysis in Stroke-Monitoring Study.
Life-threatening vaginal or systemic haemorrhage is defined as bleeding requiring >3 transfused units of blood. For the child, the incidence of stillbirth and foetal haemorrhage (intracranial or systemic) will be documented.
Ethical issues
All participating centres have to receive approval from local ethics committee and/or data protection agency before initiation of the study. A patient can be enrolled into the study after giving the informed consent, which, according to different national regulatory and legal requirements, can also be provided by a legally acceptable representative/relative or confirmed by an independent witness when the patient is not able to sign. The informed consent is required also in relation to handling of data, including access to patients’ files, sharing of data, and telephone interview. Regarding the evaluation of the newborn/infant, the informed consent from both parents will be required. Data protection will be guaranteed by the establishment of a secure web-based database (e-CRF) and appropriate patient’s data anonymisation that will be adopted also for the centralised neuroimaging analysis.
Sample size estimates
Since SiPP is an observational study, mainly with descriptive purposes, a specific sample size calculation has not been performed. Moreover, it is difficult to estimate the proportion of pregnancy/puerperium-related over total CBV events due to their rarity. As the registry gains interest from international sites, we anticipate to reach the target number of at least 250 patients prospectively recruited with pregnancy-related CBV, and 250 matched controls for each of the two groups.
Statistical analysis
Descriptive analyses will be carried out. Continuous variables will be expressed as mean (±standard deviation) or median (interquartile range) as appropriate, categorical variables will be expressed as proportions and percentages. Percentages will be calculated by dividing the number of events by the total number of patients, excluding unknown or missing values. Chi-square test or Fisher’s exact test, and Student’s t-test or Mann–Whitney U test will be used as appropriate to compare demographics, clinical and imaging characteristics between cases and controls.
For dichotomous outcome measures, chi-square test will be used for comparisons between cases and controls. For time-to-events and survival analysis, Kaplan–Meyer curves will be used to evaluate the cumulative risk of the event in both cases and controls; the comparison will be done using the log-rank test. Cox proportional hazard models will be used to compute hazard ratios after adjustment for potential confounders with a univariate p value <0.15. In the time-to-event analyses, death and withdrawal from the study (in case patients are not willing to be followed up anymore) will be considered ‘censoring events’.
Logistic regression multivariate analyses will be used to determine independent predictors of the outcome measures. Analyses for the other secondary endpoints will be carried out according to standard statistical principles for comparison of parametric or non-parametric distributions as appropriate.
Incidence and severity of adverse events, standing for safety-related events in this observational, non-interventional study, will be detailed for all patients enrolled in the study. A definitive statistical analysis plan will be written at the end of the study prior to database lock. A p value of <0.05 will be considered statistically significant.
All efforts will be made for an accurate and complete collection of information according to the study protocol for all subjects included, both cases and controls. For the handling of missing data, the last-observation-carried-forward (LOCF) method will be applied for sensitivity analyses; hence data from the previous visit will substitute missing data. In the case of missing values regarding outcome measures (death or stroke recurrence), the worst score principle will be applied. A multiple imputation technique will be also used.
Of note, prospective and retrospective data will be analysed overall and separately in order to evaluate the consistency of the results. Similarly, we will analyse data from the acute/subacute and delayed postpartum periods both overall and separately in order to better assess the risk of CBV events by the different time interval from childbirth, which is expected to be lower in the delayed postpartum period (starting from approximately six weeks and lasting up to six months).
Full study protocol will be shared with qualified parties on request to the corresponding author.
Data monitoring board – Steering Committee
The study has one Principal Investigator and one Co-Principal Investigator who are also part of the Steering Committee of the study which consists of six neurologists, members of the ESO-WISE Groups. Currently, 35 centres worldwide (mostly European) have been involved (see the Appendix for a complete list of participating centres) so far, but other centres are expressing interest to join the study. The Steering Committee’s functions include the monitoring of patient recruitment and review of trial conduct at the sites, recommendation of protocol amendments, analysis and interpretation of the study results, and preparation of the scientific publication(s).
Study organisation and funding
As pregnancy-/postpartum-related CBV events represent an uncommon condition, the largest participation will be necessary to obtain reliable and valid data. The study will be settled worldwide and participation will be promoted also through scientific societies and networks. Centres around the world interested in being involved in this project could directly contact the corresponding author or the members of the Steering Committee.
For each country, scientific coordinators are selected. The scientific coordinator promotes participation to the study on a national level.
Cases including all consecutive women admitted for a pregnancy-/postpartum-related CBV event will be prospectively recruited by participating centres. Cases will be identified among the patients admitted to stroke units/stroke centres and neurology, neurosurgery, gynaecology and intensive care wards of the different participating centres from all over Europe and outside Europe.
For each included case, a search for the matched controls will be made by the participating centres to enrol control subjects prospectively.
This organisation guarantees the feasibility of the study because the large number of centres involved will ensure to recruit the pre-specified number of cases and controls in the time period predefined for the study duration.
This study has not yet received specific grant from any funding agency in the public, commercial or not-for-profit sectors. The study will be registered in international databases (clinicaltrials.gov).
Discussion
In the management of stroke in young adult patients, the occurrence of CBV diseases during pregnancy and postpartum definitely represents a challenging situation for stroke physicians who need to carefully and continuously assess the potential risks and benefits of therapeutic interventions for both mother and foetus. Stroke in pregnancy is relatively rare, but it has been estimated a three-fold increase in incidence compared with non-pregnant women.6,7 There are many gaps in our knowledge about pregnancy-/postpartum-related stroke, including detailed understanding about etiopathogenetic mechanisms, treatments, outcomes, post-hospital discharge, and functional and female-specific outcomes. Many of the studies published on pregnancy-related stroke are based on administrative data that lack patient-level details that this registry will collect. In the next 5–10 years, it is most likely that an increasing number of pregnant women with acute ischemic stroke will be treated with intravenous thrombolysis and/or endovascular therapy with mechanical thrombectomy as new or improved imaging strategies could identify women who would benefit from reperfusion while minimising the risk to the foetus. Furthermore, in the near future, direct oral anticoagulants may be used in pregnancy, especially for women who cannot receive heparin or warfarin. More testing could be required to determine the adverse pregnancy outcomes and the risk–benefit ratio of treatments for preventing stroke and CVT during pregnancy.
For better understanding causes and outcomes of uncommon conditions like pregnancy/postpartum-related CBV events, the development of multisite, multidisciplinary registry-based studies, such as the SiPP study, is needed in order to collect an adequate number of patients, draw reliable conclusions and give definite recommendations on their management.
The originality of the SiPP study is that it is the first large-scale project trying to evaluate multiple aspects (i.e. frequency, pathophysiological mechanisms, biomolecular/genetic markers, clinical and neuroimaging characteristics, associated risk factors, types of acute interventions and secondary prevention treatments, short-term and long-term outcome of both mother and newborn, outcome predictors) of all types of CBV diseases occurring as complications in women during pregnancy/postpartum period.
The most important innovative nature of this project, compared with the other conducted so far, is in using a systematic multicentre, multi-/interdisciplinary approach to the multiple heterogeneous aspects characterising all types of CBV events potentially occurring in pregnancy/postpartum. This will allow to obtain more information on these uncommon conditions and mostly unmet clinical needs and will provide a better insight into their biology. Information from the SiPP study will be relevant to support the management of CBV events occurring during pregnancy and postpartum. Data will be available upon reasonable request to other investigators for nested studies. This may further promote advances in the field.
The search for biomolecular/genetic markers and outcome predictors in the SiPP study will allow to individuate specific targets for the development of novel treatments, new surrogate biomarkers, and to build a more complete outcome predictive model, and hence a clinical-radiological and biomolecular score, in order to better optimise the management of CBV diseases associated with pregnancy/postpartum, in terms of risk stratification. The prospect will be that of developing a personalised medicine enabling clinicians to provide the patients with the most appropriate treatment. Therefore, the results of this international, multicentre, multidisciplinary study could have an extraordinary scientific, clinical and socio-economic impact.
Summary and conclusions
The SiPP study, supported by the ESO-WISE group, has been designed, by using a multidisciplinary approach, to evaluate the characteristics and management of CBV events in pregnant/postpartum women, to clarify pathophysiological mechanisms underlying the occurrence of these events including biomolecular aspects and to assess the short- and long-term CBV and global cardiovascular outcomes of these patients, their predictors and foetus/infant outcome.
Supplemental Material
ESO893512 Supplemental Material1 - Supplemental material for SiPP (Stroke in Pregnancy and Postpartum): A prospective, observational, international, multicentre study on pathophysiological mechanisms, clinical profile, management and outcome of cerebrovascular diseases in pregnant and postpartum women
Supplemental material, ESO893512 Supplemental Material1 for SiPP (Stroke in Pregnancy and Postpartum): A prospective, observational, international, multicentre study on pathophysiological mechanisms, clinical profile, management and outcome of cerebrovascular diseases in pregnant and postpartum women by Svetlana Lorenzano, Christine Kremer, Aleksandra Pavlovic, Dejana R Jovanovic, Else Charlotte Sandset, Hanne Christensen, Cheryl Bushnell, Anita Arsovska, Nikola Sprigg, Christine Roffe, Petra Ijäs, Zuzana Gdovinova, Anne Alexandrov, Marialuisa Zedde, Rossana Tassi, Monica Acciaresi, Maria Lantz, Katharina Sunnerhagen, Marija Zarkov, Kirsi Rantanen, Fabienne Perren, Helle K Iversen, Christina Kruuse, Agnieszka Slowik, Paola Palazzo, Janika Korv, Annette Fromm, Arijana Lovrencic-Huzjan, Eleni Korompoki, Ana Catarina Fonseca, Seana L Gall, Freimuth Brunner, Valeria Caso, Simona Sacco and for the SiPP Trial Investigators and ESO-WISE Group in European Stroke Journal
Supplemental Material
ESO893512 Supplemental Material2 - Supplemental material for SiPP (Stroke in Pregnancy and Postpartum): A prospective, observational, international, multicentre study on pathophysiological mechanisms, clinical profile, management and outcome of cerebrovascular diseases in pregnant and postpartum women
Supplemental material, ESO893512 Supplemental Material2 for SiPP (Stroke in Pregnancy and Postpartum): A prospective, observational, international, multicentre study on pathophysiological mechanisms, clinical profile, management and outcome of cerebrovascular diseases in pregnant and postpartum women by Svetlana Lorenzano, Christine Kremer, Aleksandra Pavlovic, Dejana R Jovanovic, Else Charlotte Sandset, Hanne Christensen, Cheryl Bushnell, Anita Arsovska, Nikola Sprigg, Christine Roffe, Petra Ijäs, Zuzana Gdovinova, Anne Alexandrov, Marialuisa Zedde, Rossana Tassi, Monica Acciaresi, Maria Lantz, Katharina Sunnerhagen, Marija Zarkov, Kirsi Rantanen, Fabienne Perren, Helle K Iversen, Christina Kruuse, Agnieszka Slowik, Paola Palazzo, Janika Korv, Annette Fromm, Arijana Lovrencic-Huzjan, Eleni Korompoki, Ana Catarina Fonseca, Seana L Gall, Freimuth Brunner, Valeria Caso, Simona Sacco and for the SiPP Trial Investigators and ESO-WISE Group in European Stroke Journal
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Svetlana Lorenzano served as expert consultant for Boehringer Ingelheim from 2013 to 2014; she received two travel grants from Boehringer Ingelheim (one in 2016 one in 2017), one travel grant from Bayer (2014), Quintiles IMS (2017), and Daichii Sankyo (2017) for meetings and conferences. All the remaining authors declare that there is no conflict of interest.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
A patient can be enrolled into the study after giving his/her informed consent. Regarding the evaluation of the newborn/infant, the informed consent from both parents is required.
Ethical approval
All participating centres have to receive approval from local ethics committee and/or data protection agency before initiation of the study.
Contributorship
SL conceptualised and designed the study, drafted the study protocol, drafted the article and revised the article critically for important intellectual content. SS conceptualised and designed the study, reviewed the study protocol and revised the article critically for important intellectual content. All the remaining co-authors revised the article critically for important intellectual content.
Guarantor
SL as first author and SS as one of the two senior authors.
Acknowledgements
The authors thank all the members of the ESO-WISE Group for their support. The authors also thank Prof. Danilo Toni and Dr. Ales Tomek for their interest in participating in this study.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.