
Abstract
Background
The cause of cervical artery dissection is not well understood. We test the hypothesis that mutations in genes associated with known arterial connective tissue disorders are enriched in patients with familial cervical artery dissection.
Patients and methods
Patient duos from nine pedigrees with familial cervical artery dissection were analyzed by whole exome sequencing. Single nucleotide variants in a panel of 11 candidate genes (ACTA2, MYH11, FBN1, TGFBR1, TGFBR2, TGFB2, COL3A1, COL4A1, SMAD3, MYLK and SLC2A10) were prioritized according to functionality (stop-loss, nonsense, and missense variants with polyphen-2 score ≥0.95). Variants classified as “benign” or “likely benign” in the ClinVar database were excluded from further analysis. For comparison, non-benign stop-loss, nonsense and missense variants with polyphen-2 score ≥0.95 in the same panel of candidate genes were identified in the European non-Finnish population of the ExAC database (n = 33,370).
Results
Non-benign Single nucleotide variants in both affected patients were identified in four of the nine cervical artery dissection families (COL3A1; Gly324Ser, FBN1: Arg2554Trp, COL4A1: Pro116Leu, and TGFBR2: Ala292Thr) yielding an allele frequency of 22.2% (4/18). In the comparison group, 1782 variants were present in 33,370 subjects from the ExAC database (allele frequency: 1782/66,740 = 2.7%; p = 0.0008; odds ratio = 14.2; 95% confidence interval = 3.8–52.9).
Conclusion
Cervical artery dissection families showed enrichment for non-benign variants in genes associated with arterial connective tissue disorders. The observation that findings differed across families indicates genetic heterogeneity of familial cervical artery dissection.
Keywords
Spontaneous dissection of the cervical arteries (CeAD) is an important cause of ischemic stroke in younger adults. The detailed causes leading to CeAD are not well understood.1–3 Major trauma of the neck or head is a rare cause of CeAD, but minor mechanical trigger events were reported in less than half of the patients, 4 which are otherwise healthy individuals typically lacking atherosclerotic vascular risk factors.4,5 Genetic factors were assumed to modify the risk of occurrence of CeAD. 6 Indeed, carriers of the major allele of a common variant (rs9349379) of the phosphatase and actin regulator 1 encoding gene (PHACTR1) were recently found to have a slightly reduced risk for CeAD. 7 Clinical connective tissue signs and electron microscopic connective tissue alterations were associated with CeAD.8,9 Nevertheless, known inherited connective tissue disorders seemed exceptional with a frequency of <1% in a recent large series of patients.10,11 Moreover, mutation search in few candidate genes associated with inherited connective tissue disorders lead to isolated suggestive findings, but was unsuccessful in most patients.8,12–14 Thus, additional genetic factors modifying the risk for CeAD are yet to be discovered.
In the current study, we assumed that unrecognized manifestations of known inherited connective tissue disorders may predispose to CeAD. To test this assumption, we considered familial CeAD lacking clinically apparent connective tissue disorders as ideal model. On the basis of published genetic studies of arterial connective tissue disorders, we performed a next generation sequencing study and defined a panel of 11 candidate genes which were associated with arterial connective tissue disorders.15–18 In nine families with familial CeAD, we searched for putative disease-associated, deleterious variants in the predefined panel of candidate genes. Findings were compared with published findings from the European non-Finnish population of the ExAC database (http://exac.broadinstitute.org/).19,20
Material and methods
CeAD patients with a family history of CeAD were identified between 2004 and 2015 in databases of neurology departments and stroke centers with a special interest in CeAD.21–22 All identified families were of German or Swiss–German origin. All affected patients were examined, diagnosed, and treated by stroke neurologists. The diagnosis of CeAD was verified and based on arterial imaging, in particular on magnetic resonance imaging findings. 2 This includes the presence of at least one of the following criteria: mural hematoma, aneurysmal dilatation, long tapering stenosis, intimal flap, double lumen, or occlusion ≥2 cm above the carotid bifurcation revealing an aneurysmal dilatation or a long tapering stenosis after recanalization in a cervical artery (i.e. internal carotid artery (ICA) or vertebral artery (VA)). For the current analysis, the following standardized variables were analyzed, applying criteria used in previous publications: 5 age (at onset), sex, site of dissection (ICA orVA), side, type of familial relationship and country of birth.
Peripheral blood was used for DNA extraction. Exome sequencing was performed at the German Research Center for Environmental Health, Helmholtz Zentrum München, on a Genome Analyzer IIx system (Illumina) after in-solution enrichment of exonic sequences (SureSelect Human All Exon 38 Mb kit, Agilent). Read alignment was performed with BWA (version 0.5.8) to the human genome assembly hg19. Single-nucleotide variants (SNVs) were detected with SAMtools (v 0.1.7).
On the basis of published studies,15–18 we defined a panel of 11 candidate genes (ACTA2, MYH11, FBN1, COL3A1, COL4A1, TGFBR1, TGFBR2, TGFB2, SMAD3, MYLK, SLC2A10) associated with arterial connective tissue disorders (vascular Ehlers-Danlos syndrome, Marfan syndrome, Loeys-Dietz syndrome, familial thoracic aortic aneurysms and dissections, arterial tortuosity syndrome). SNV findings from the CeAD families were prioritized if they (1) had a coverage (depth) of ≥40 reads; (2) caused nonsense or stop-loss substitutions in the encoded gene product, or missense substitutions with polyphen-2 probability scores ≥0.95. In a final filtering step, prioritized missense mutations with polyphen-2 scores ≥0.95 that were classified as “benign”, “likely benign” or “benign/likely benign” 23 in the ClinVar browser (https://www.ncbi.nlm.nih.gov/clinvar/) 24 were removed from subsequent analysis.
Prioritized “non-benign” SNV findings in the CeAD families were compared with reported findings in the European (non-Finnish) populations from the ExAC database (http://exac.broadinstitute.org/).19,20 For each of the 11 arterial connective tissue genes from the candidate panel, all SNVs causing nonsense or stop-loss substitutions or causing missense substitution with polyphen-2 proability score ≥0.95 and not classified as “benign,” “likely benign,” or “benign/likely benign” in ClinVar were looked up in the ExAC Browser and scored as non-benign variants occurring in the population.
Results
The patient population, comprising 18 affected first-degree relatives from nine families.
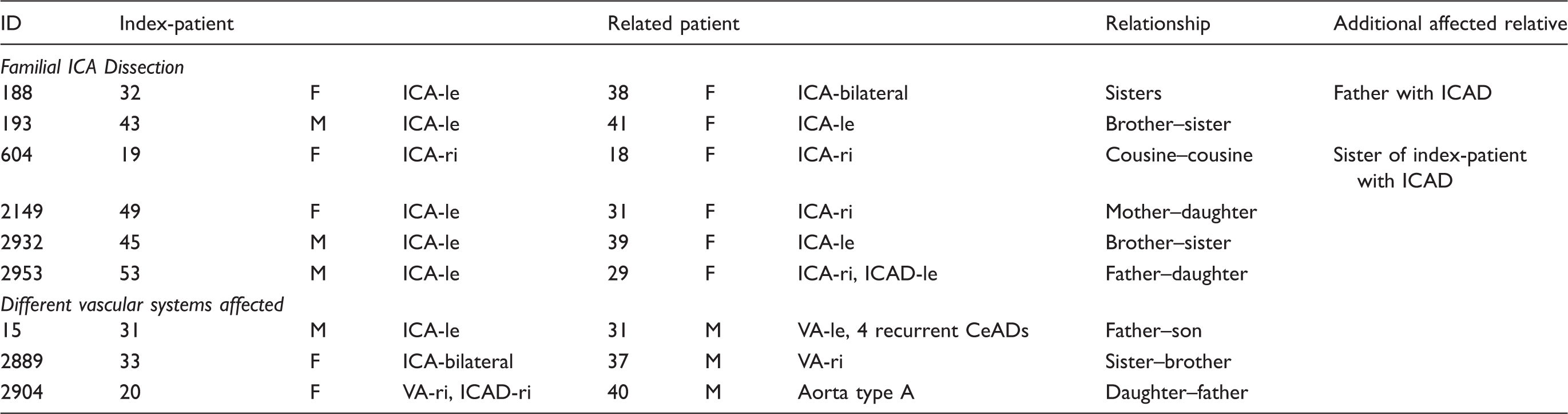
Note: Numbers indicate years of age at onset of first cervical artery dissection (CeAD) event. Family 2953 was of German–Swiss origin, all other families were from German origin. F: female; M: male; VA: vertebral artery; ICA: internal carotid artery; le: left; ri: right.
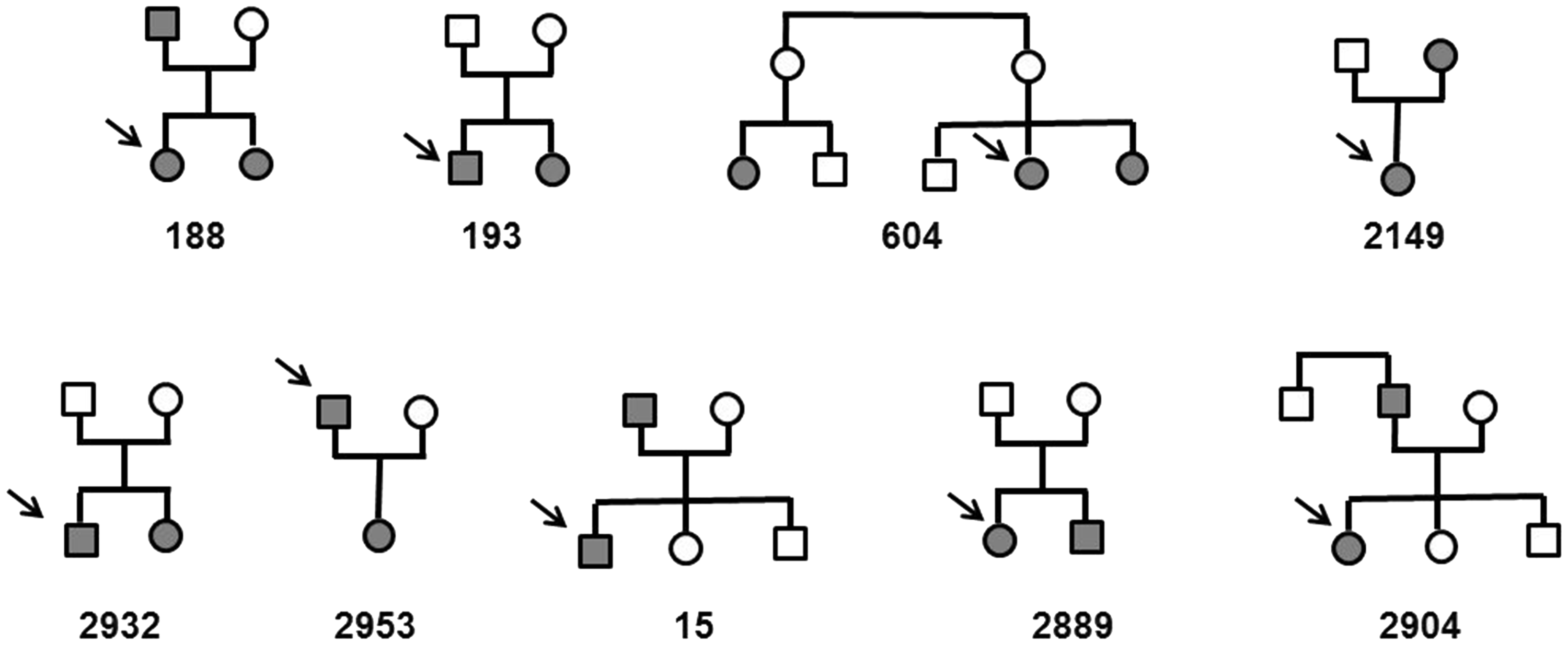
Pedigrees of the analysed families. Arrows indicate index-patients (index patients of Table 1). Filled symbols indicate patients with CeAD, open symbols indicate relatives with documented self-reported absence of CeAD. Genetic analysis was performed solely in affected relatives.
SNV were considered as deleterious, if they (1) caused a premature stop of the encoded gene product, (2) removed a stop-codon (“stop-loss”) or (3) induced a missense mutation with polyphen-2 probability score ≥0.95 and with ClinVar classification other than “benign” or “non-benign”. A total of 1242 SNV in the panel of candidate genes were identified in the patient sample, among them 142 SNVs leading to nonsense, stop-loss or missense substitution. Nine missense mutations with polyphen-2 probability score ≥0.95 were classified as “non-benign”. These non-benign variants in genes associated with arterial connective tissue disorders occurred in four of the nine analyzed CeAD families. These include mutations in COL3A1, FBN1, COL4A1, and TGFBR2, each exclusively in one family. Analysis of copy number variation in the next generation sequencing data did not indicate the occurrence of variants larger than five exons in any of the cases (data not shown).
Prioritized non-benign variants in the patient sample.
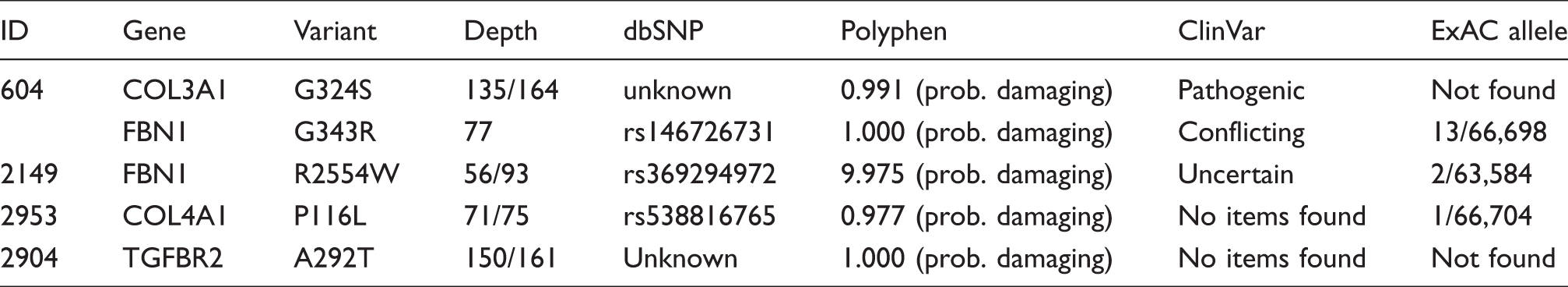
Depth: coverage for both affected relatives or (in case of the FBN G343R variant) for a single patient; polyphen: polyphen-2 probability score (and assigned classification); prob. damaging: probably damaging; ClinVar: classification of the variant according to the ClinVar database; conflicting: Conflicting interpretations of pathogenicity; uncertain: uncertain significance; ExAC allele frequency: observed allele frequency in the non-Finnish European populations of the ExAC database.
SNVs in the candidate gene panel identified in CeAD patients and in European control subjects.
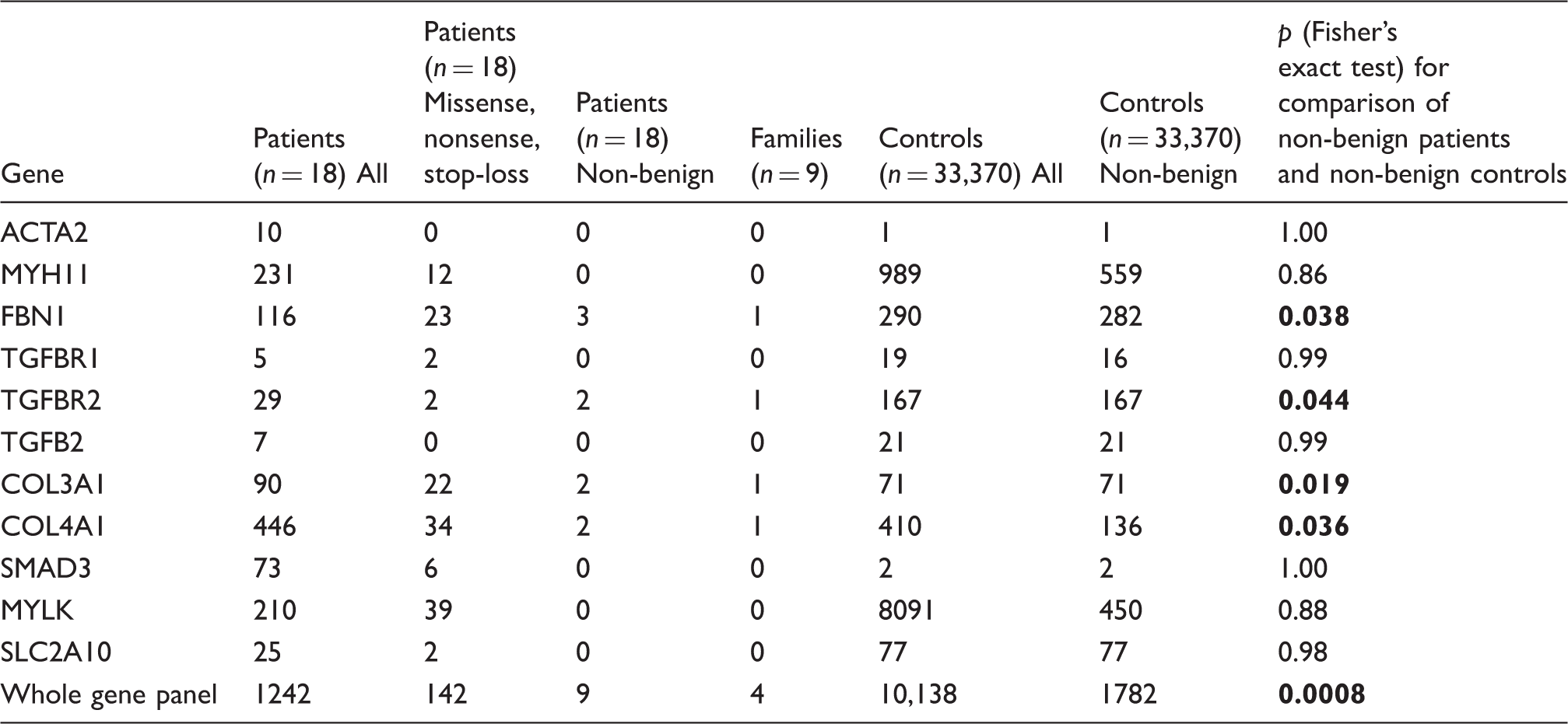
Note: For the patient sample, columns show all identified exome variants (including synonymous variants, splice variants as well as intronic, 3′ and 5′ UTR variants), all nonsense, stop-loss and missense variants and (third column), nonsense, stop-loss variants as well as missense variants with polyphen-2 probability scores ≥0.95 and with ClinVar classification other than “benign” or likely “benign”. The prioritized variants that were present in both affected relatives of a family were considered as familial. All European non-Finnish carriers of a nonsense or stop-loss variant or of a missense variant with polyphen-2 probability scores ≥0.96 were counted for each of the 11 candidate genes. In a final prioritization step, variants that were classified as “benign” or “likely benign” were not analyzed further.
The finding of four non-benign SNPs in nine families (allele frequency: 4/18 = 22.2%), compared to 1782 findings in 33,370 population controls (allele frequency: 1782/66,740 = 2.7%) suggested that carriers with non-benign SNPs in genes associated with arterial connective tissue disorders were at increased risk for familial CeAD (Table 3; p = 0.0008; odds ratio = 14.2; 95% confidence interval = 3.8–52.9).
Discussion
This analysis of rare deleterious SNVs in a predefined set of candidate genes had the following key findings: (1) non-benign variants in genes associated with inherited arterial connective tissue disorders were observed at low prevalence in the European non-Finnish population, but were about 10 times more frequent in patients with familial CeAD; (2) our study did not identify a single CeAD-specific candidate gene, but observed a polygenic burden of variants across different genes associated with known inherited connective tissue disorders, suggesting genetic heterogeneity of the CeAD phenotype.
Mutations in COL3A1 or TGFBR2 in CeAD patients had been reported in anecdotal case reports,7,13,14 whereas this had been not the case for FBN1 and COL4A1. The FBN1 Arg2554Trp mutation in pedigree 2149 was described before in three family members with atypical Marfan syndrome affecting the cardiovascular system, but neither the eyes nor the skeleton. 25 Recently, we found this same Arg2544Trp missense mutation of FBN1 in a young woman with multiple CeAD and arachnoidactily (Baumgartner RW and Grond-Ginsbach C, unpublished data). Mutations in COL4A1 were associated with a broad spectrum of symptoms (HANAC (dominant hereditary angiopathy with nephropathy, aneurysms, and muscle cramps) syndrome), including aneurysms, small vessel disease and hemorrhagic stroke. 26 The affected father of family 2953 developed a large dilatative ICA aneurysm secondary to his ICAD, which may be considered as sign of an underlying syndrome. 18
No mutations were identified in the remaining four families. Other types of genetic variation may play a role, including small deletions or duplications (indels) causing frame-shift mutations or large structural variant like a deletion covering the whole COL3A1 and COL5A2 genes that was identified in a pilot study of copy number variation (CNV) in CeAD patients. 14 Interestingly, a recent follow-up CNV study in a large patients’ sample associated CeAD with genetic variants affecting the development of the vascular system and found such variants in both affected sibs of a family. 27 The lack of findings in five families may also indicate that the current panel of 11 candidate genes was too selective or that the cut-off for the prioritization of variants (with polyphen-2 probability scores ≥0.95 and with non-benign ClinVar classification) might have been too stringent.
Familial occurrence of CeAD is rare (i.e.<1%).11,28,29 Hence, the presented sample of nine affected duos with familial CeAD, all white Caucasians from Germany or from the German speaking part of Switzerland, provided a unique material for a genetic analysis. The analysis of a large group of unrelated subjects for the ExAC database for comparison was a further strength of this study. However, we had no information on age, sex or health state of the analyzed ExAC controls. Nevertheless, our study has several limitations: The study sample of patients was small, which reduced the likelihood of recurrent findings in different families. Moreover, the study sample was highly selective, as most CeAD patients are sporadic (i.e. not familial) and familial occurrence of CeAD is in fact exceptional. Sequence analysis of larger series of patients is therefore needed to estimate the contribution of rare genetic variants in the pathogenesis of sporadic CeAD. The variants in this study were prioritized with regard to functionality, but not with regard to frequency. As all prioritized familial variants appeared to be extremely rare in the non-Finnish European controls, it is tempting to reanalyze the frequency of non-benign variants in the control population after exclusion of common variants. Prioritization of rare variants in the ExAC sample would suggest an even more dramatic enrichment of variants in the familial CeAD patients (data not shown).
Conclusion
CeAD families showed enrichment for deleterious variants in genes associated with arterial connective tissue disorders. The observation that findings were identical within each family but different across families indicates genetic heterogeneity of CeAD.
Footnotes
Acknowledgements
The authors thank Werner Hacke for continuous support, Dr Tim Strom for valuable advice, and Inge Werner for excellent technical assistance.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: MD received funding from the Vascular Dementia Research Foundation; this research was partly supported by the Basel Stroke Funds and the Neurological Clinic of the University Hospital Basel.
Ethical approval
The study protocol was approved by relevant local authorities in all participating centers and complied with all national regulations concerning ethics committee approval and informed consent.
Informed Consent
All patients gave written informed consent prior to study participation
Guarantor
CG-G.
Contributorship
CGG, MD, and STE conceived the study. TB, MK, PL, CT, PE, JJM, AA, AS, GRF, AT, JM, and RWB were involved in patient recruitment. CGG, SSA, PE, and STE interpreted the data and wrote the manuscript. All authors reviewed and edited the manuscript and approved the final version.