
Abstract
Von Recklinghausen disease is the most common phacomatosis. It can affect many systems, including the bone system. Through these 2 cases, we illustrate the bone manifestations of this disease.
Introduction
Neurofibromatosis type 1 is a genetic disease with variable manifestations.
Its diagnosis is largely clinical with well-defined criteria; in some cases, it is supported by imaging which notes peculiar anomalies.
It usually manifests itself in adolescence with dermatological signs in the foreground. In rare cases, the disease is revealed by bone manifestations.
Observation 1
The patient was an 8-year year-old boy, the first of 2 siblings, who was referred to us for exploration of a left lower limb deformity dating back to birth.
The interrogation underlined a notion of a fall from his height at the age of 1 year; it did not find a family context, infectious, or orthopedic surgery.
The clinical examination showed well-circumscribed brown stains of more than 5 mm on the trunk and limbs (Figure 1), and the rest of the clinical examination, in particular neurological, ophthalmological and ENT, was without particularities. An X-ray of the left lower limb was performed (Figure 2), followed by one of the spine (Figure 3). Additional MRI and bone marrow studies showed (Figures 3 and 4).

Anterior incurvation of the left leg with brown stains on the lateral aspect of the back and the medial aspect of the left leg.

(A) Antero-medial incurvation of the tibia, angulation of the fibula with densification and cortical thickening reducing the medullary canal and solution of bony continuity associated with a callus, in relation to a congenital pseudarthrosis. (B) Respect for spinal statics, posterior scalloping more marked at the level of the vertebral body of L5.
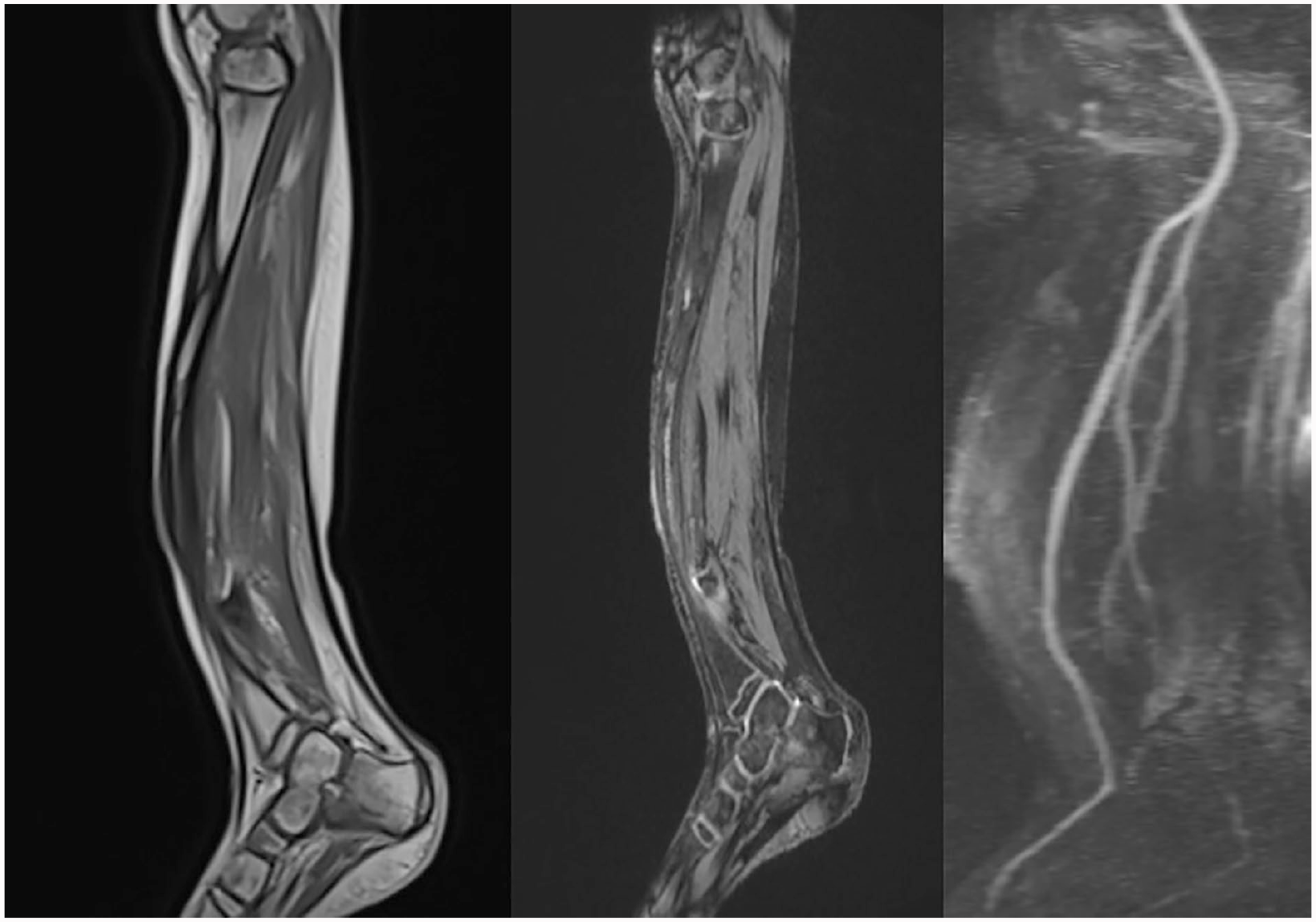
Absence of vascular anomalies, bone signal anomaly with tibio-peroneal pseudarthrosis, absence of soft tissue proliferation.
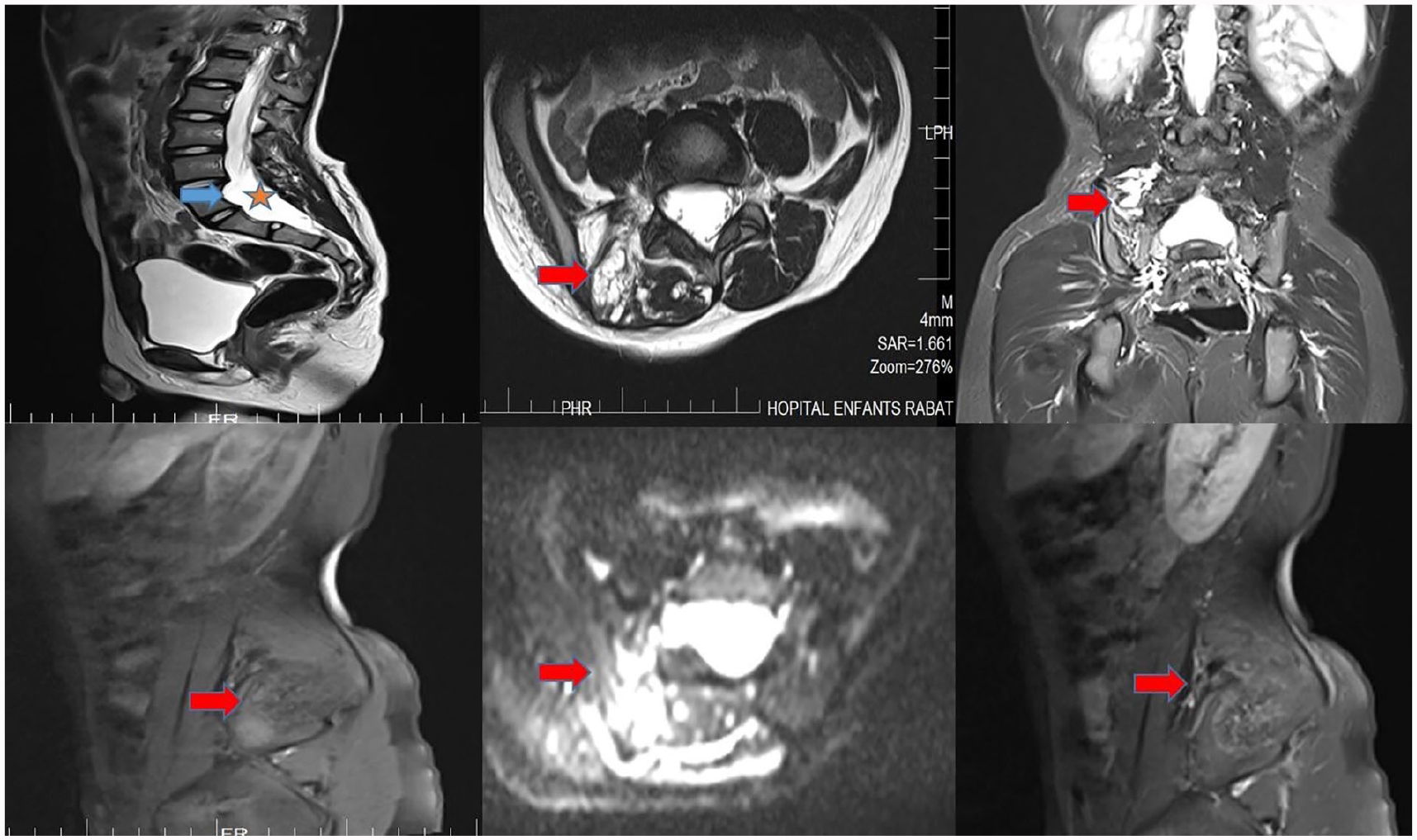
Multiplanar T2 MRI sections, T1 sag without gado, ax DWI, and T1 sag with injection showing dural ectasia with posterior scalloping of the vertebral bodies of L5 and S1. At the level of the lumbosacral vertebrae (L4, L5, and S1), presence of several right paraspinal serpiginous formations enlarging the homolateral foramen, with indistinct contours pushing back the right spinal muscles and the homolateral psoas of heterogeneous hyper T2 signal centered by a hypo signal zone, hypoT1 weakly enhanced after injection of gado in connection with plexiform neuromas.
Observation 2
A 17-year-old patient with no previous history, presented with a royal tumor of the left hemiface. The clinical examination revealed a facial deformity with multiple brown stains over 15 mm, the number of which exceeded 6. The rest of the somatic examination was unremarkable. A brain CT scan was performed (Figure 5).

Sphenoidal dysplasia, marked by the absence of the large left wing of the sphenoid.
Discussion
Neurofibromatosis type 1, known as Von Recklinghausen disease, is the most common phacomatosis (1/3000 births). 1 It is diagnosed on the basis of a number of well-defined criteria, including clinical and imaging.
These criteria include the presence of at least (6 brown spots, 2 neurofibromas or 1 plexiform neuroma, 2 Lisch nodules) the presence of axillary or inguinal lentigines, a glioma of the optic chiasm, a characteristic bone lesion and a first degree family history. The diagnosis is made when at least 2 of these criteria are present. 2
The role of imaging in this condition is essentially to search for neurological, bone or vascular damage; this is made possible by conventional radiology and cross-sectional imaging (CT/MRI). Thus, at the bone level, the anomalies found may concern the axial or appendicular skeleton with areas of predilection such as3,4:
-The skull in the form of sphenoid dysplasia (absence of the small or large wing of the sphenoid), bony defect opposite the lambdoid suture, mastoid, maxillary or ethmoidal hypoplasia, granular calcifications along the temporal horn or macrocrania outside of any hydrocephalus .
-The rachis or cyphoscoliosis or scoliosis may be encountered, as well as costal anomalies (small, wavy, upturned, eroded or destroyed appearance), vertebral anomalies (anterior, lateral or posterior scalloping of the vertebral bodies, thinning of the pedicles or transverse processes, enlargement of the intervertebral foramina and the spinal canal, frequency of vertebral malformations). It is sometimes associated with a dural ectasia, a meningocele or nerve damage.
-The long bones, marked by, the accentuation of the curvature of the long bones in particular of the tibia at the level of the junction 1/3 medium-1/3 inferior of the diaphysis and the anomaly of its texture (thinning diameter, pseudo-cystic or sclerotic osteolytic reorganizations) The patient may have a hypoplastic or snake-like fibula, fractures and pseudoarthroses of the tibia or, more rarely, of the fibula, ulna or radius, large subperiosteal haematomas, focal bone lesions related to erosion by nerve tumors or non-ossifying fibroids, as well as joint damage (pinching of the interline and/or marginal bone erosions).
In addition to these bone lesions, there are dural, nerve, and vascular lesions such as neurofibromas, hemangiomatoses, lymphangiomatoses, dural ectasias and even meningoceles that are well studied by magnetic resonance imaging. MRI allows on the one hand to characterize and map these lesions, to appreciate the bone vascularization in case of pseudarthrosis, and on the other hand the monitoring of neurofibromas where the risk of degeneration exists. 4
Whatever the form in which it appears, the approach to this disease must be global for a complete lesion assessment and better management.
Conclusion
Neurofibromatosis type 1 is a genetic disease with a predominantly cutaneous, vasculo-nerve and bone tropism. Pseudarthrosis of the tibia is one of the bone manifestations of this disease and can be revealing.
Footnotes
Author Contributions
All authors contributed equally to this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.