
Abstract
Anorectal malformations are relatively common anomalies encountered in pediatric surgical practice. They are usually recognized at birth with absent anal canal or failure to pass meconium and hence can lead to life threatening bowel obstruction without surgical intervention. We are reporting a rare case of non-syndromic, sporadic, terminal colovesical fistula associated with high anorectal malformation not associated with congenital pouch colon in a neonate.
Keywords
Introduction
Anorectal malformations (ARM) have an incidence of 1 in 1:2500 to 1:1500 live births with a mild male predominance and have a variable clinical presentations ranging from mild forms that might require only minor surgical interventions to more complicated cases that need to be managed with multi-staged operations. Though the cause of ARM is unknown, the arrest of the descent of the urorectal septum toward the cloacal membrane between the 4th and 8th weeks of gestation has been considered the basic event leading to ARM.
Case Report
A 2-hours-old male neonate born by full term cesarean section by non-consanguineously married couple as a first child. Baby had a birth weight of 3 kg was referred to s with absence of anal orifice (Figure 1A). On admission baby was stable hemodynamic ally with normal tone and activity having good cry and sucking. Baby’s length was 49 cm, head circumference 35 cm and weight of 2.95 kgs.We evaluated the baby with routine blood investigations, infantogram and Prone cross-table lateral shot through radiography revaluated it to be high anorectal malformation (Figure 1B, C); distal bowel gas shadow ending above pubo coccygeal (PC) line, hence high sigmoid colostomy was done after 10 hours of birth (Figure 1D).
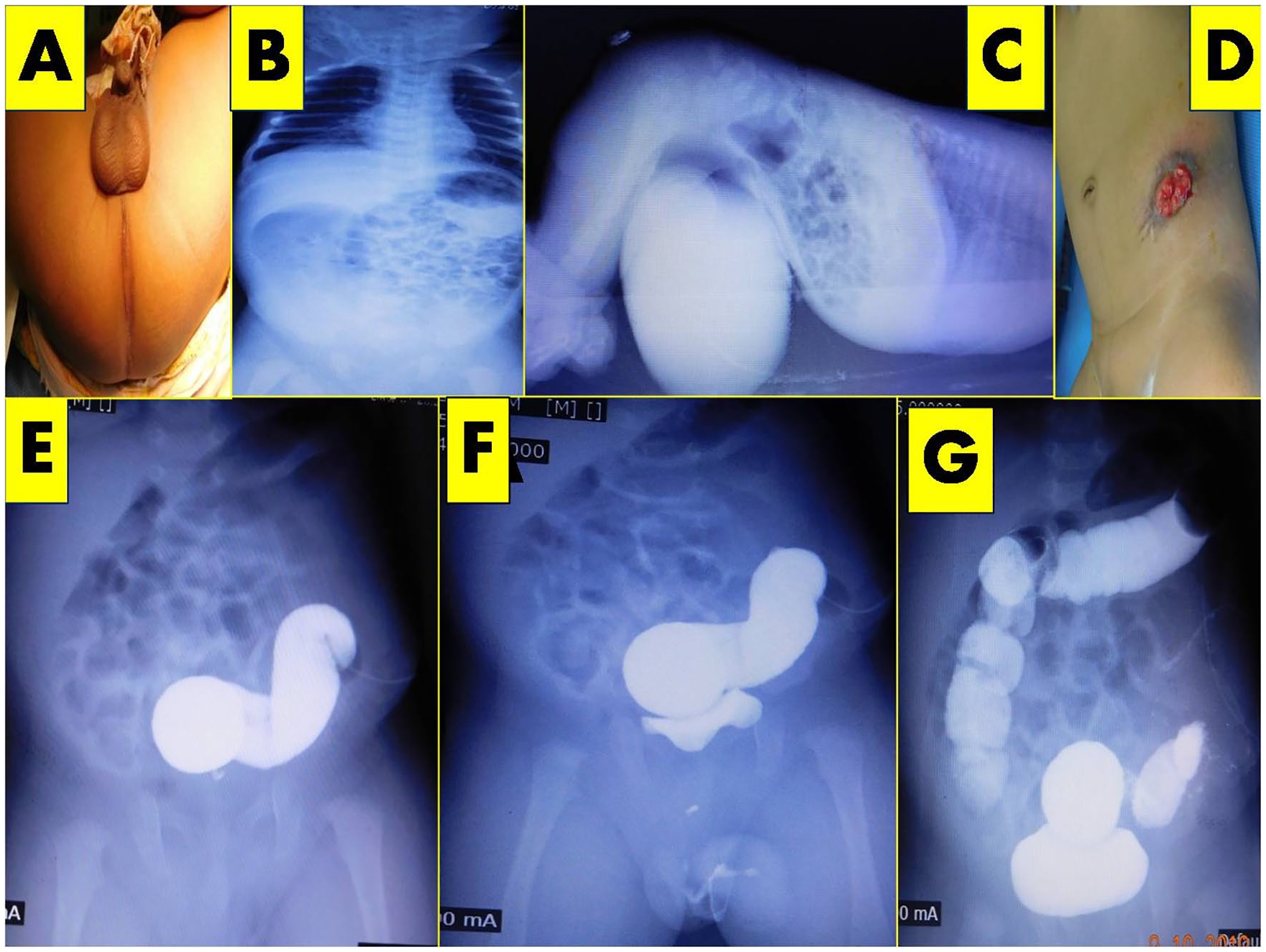
(A) Clinical appearance of ARM. (B) Infantogram. (C) Prone table lateral shoot through radiography showing distal bowel air shadow above PC line. (D) High sigmoid colostomy. (E and F) pressure augmented distal colography image showing a terminal colo vesical fistula.
Baby was doing well on follow up having normal stoma output and was passing urine normally. Routine ultrasonography, 2d echocardiography, endocrinological, genetic evaluation as well as spine examination were essentially normal.
Surprisingly, author found the presence of a terminal colo vesical fistula on electively planned distal loopgraphy, the non-dilated colon having normal caliber, width as well as length connecting to the normal size bladder having normal upper urinary tracts (Figure 1E–G). Baby underwent abdominoperinael pull though at the age of 1.5months; intra operatively on posterior sagittal approach, there was no distal bowel except the bladder. Hence tract was created in posteror sagital region and sphincter muscle complex was closed around the red rubber tube (Figure 2A, B). Abdomen opened through Pfannesteil incision where a normal redundant sigmoid bowel ending as a wide fistula, the terminal colovesical fistula of 2 cm in length and 05 cm width into the posterior wall bladder between dome and trigone without any trace of distal bowel or rectum (Figure 2C, D). The terminal colo vesical fistula was disconnected from the posterior wall of the bladder, and sigmoid colon was brought to perineum through previously made posterior sagitaal incision along the red rubber tube and anoplasty was done (Figure 2E). Histopathological examination of the terminal end of colon was essentially normal with ganglionic innervations. Colostomy closure was done after 2 months of age once the paerineal wound healed. Baby is now three years old and passing normal stolls via neoanus uneventfully (Figure 2E, F).

(A) PSARP approach showing only bladder without any bowel or fistula, forceps pointing at posterior wall of bladder. (B) PSARP wound closed over red rubber tube. (C) Blue arrow pointing at – terminal colovesical fistula; sigmoid colon ending as a fistula in the posterior wall of the bladder between dome and trigone. (D) Opened terminal colovesical fistula over an infant feeding tube. (E) Anoplasty. (F) Colostomy closure.
Discussion
The anorectal malformations occur in all ethnic groups, ranging in severity from relatively mild defects such as anteriorly displaced anus, covered anal stenosis, to more severe complete anal agenesis and rectal atresia. Low anorectal lesions being more common than high or intermediate lesions. 1
Most of these anomalies are detected at birth by visual inspection or with failure to pass meconium. Alternatively, meconium may be passed via a vaginal, urethral, or perineal fitula.
There is remarkably higher preponderance in boys for more complex ARMs, whereas the less severe types such as perineal or vestibular fistulae have been reported more frequently in girls.1,2
The internal anatomy is predicted by the clinical examination, “invertogram” radiology, the presence of gas on radiology in other viscera, radiopaque contrast studies of the fistula, urinary tract or the bowel, ultrasound examination of the abdomen, pelvis and spine, computed tomography (CT) and magnetic resonance imaging (MRI), and finally the occasional use of endoscop.1 –4
Surgical treatment for anorectal malformations, the low lesions are treated in neonatal period with excellent prognosis. The high and intermediate ARM requires initial colostomy followed by staged reconstruction.1 –4
Associated anomalies are more common in boys (52-63%) and the higher the ARM, the higher the risk of associated anomalies, commonly ranging from 44% to 67%. Associated anomalies can be twice as prevalent in patients with higher anomalies as in those with lower lesions.1 –4 Our baby did not have any associated anomalies.
As per the literature the clovesical fistulae are common in association with congenital pouch colon. Only one case of non-terminal colovesical fistula associated with high ARM has been reported in the literature. 5
Author had a 2 hours old male neonate presenting with absence of anal orifice; radiography revealed the supra levator lesion above PC line, hence initially high sigmoid colostomy was done. However, surprisingly when pressure augmented distal colography after 1 month performed, revealed a terminal colovesical fisluta without distal bowel (rectum). Abdomino perineal pull though was performed; terminal colovesical disconnected from the posterior wall of the bladder at 1.5 months, and colostomy closure was done at the age of 2 months successfully.
Author is sharing her experience of managing a unique first ever case of non-syndromic, sporadic, terminal colovesical fistula associated with high anorectal malformation without having congenital pouch colon or any other co existent anomalies. This is the first ever case of staged surgical procedures to be performed earliest in infantile period for a high ARM.
Footnotes
Acknowledgements
Authors would like to thank all her pediatric surgical colleagues, anesthetists, OT staffs, radiology technicians, radiologists and IT Department and MR Department of IGICH, Bangalore for their kind support and encouragement.
Author’s Note
This article does not involve human participants or animals. Informed consent has been taken from the parents regarding the publication article as well as for photography. Signed consent form has been attached. Author herself, the operating surgeon has contributed to the article.
Author Contributions
Jayalaxmi Shripati Aihole: The operating surgeon; concept and designed the study, analyzed data and drafted the manuscript; collected the data and data analysis.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Taken from institutional committee IGICH000234.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
Taken from parents.