
Abstract

Introduction
Cystinuria (OMIM 220100) is characterized by the defective reabsorption of cystine, lysine, ornithine, and arginine in the brush border membrane of the proximal renal tubule and the defective absorption of these amino acid in the epithelial cells of the gastrointestinal tract. High urinary cystine concentrations may cause the formation of recurring renal stones. Mutations in 2 genes, SLC3A1 (rBAT) and SLC7A9 (BAT1), are known to be responsible for this disease.1,2 In this article, we report the case of a 4-year-old girl with cystinuria caused by a heterozygous novel mutation in SLC3A1.
Case Report
The patient was a healthy 4-year-old Japanese girl, who was the second child of healthy non-consanguineous parents. She was referred to our hospital because of elevated serum liver enzyme levels of unknown cause, which were discovered by chance. Her blood urine nitrogen level was 18.4 mg/dL, and her serum creatinine level was 0.3 mg/dL. Urinary sediments contained 50 to 99 erythrocytes per high-power field. An abdominal X-ray image showed a staghorn calculus in the left kidney. Intravenous pyelography detected left duplicated renal pelvis. Urinary concentrations of cystine, ornithine, lysine, and arginine were 1078.2, 961.0, 3646.2, and 1728.7 µmol/day, respectively. Renal ultrasonography showed hyperechoic lesions with sonic shadows in the left renal pelvis, as shown in Figure 1a, and hexagonal crystals were detected in her urine sediment, as shown in Figure 1b. These findings led to the diagnosis of cystinuria. Elevated liver enzyme levels were unlikely related to cystinuria because they improved without any treatment. Treatment with tiopronin and citrate was initiated for cystinuria, and she was regularly followed-up at our hospital. Although X-ray findings of urolithiasis did not improve as shown in Figure 1c, she did not suffer from flank pain.
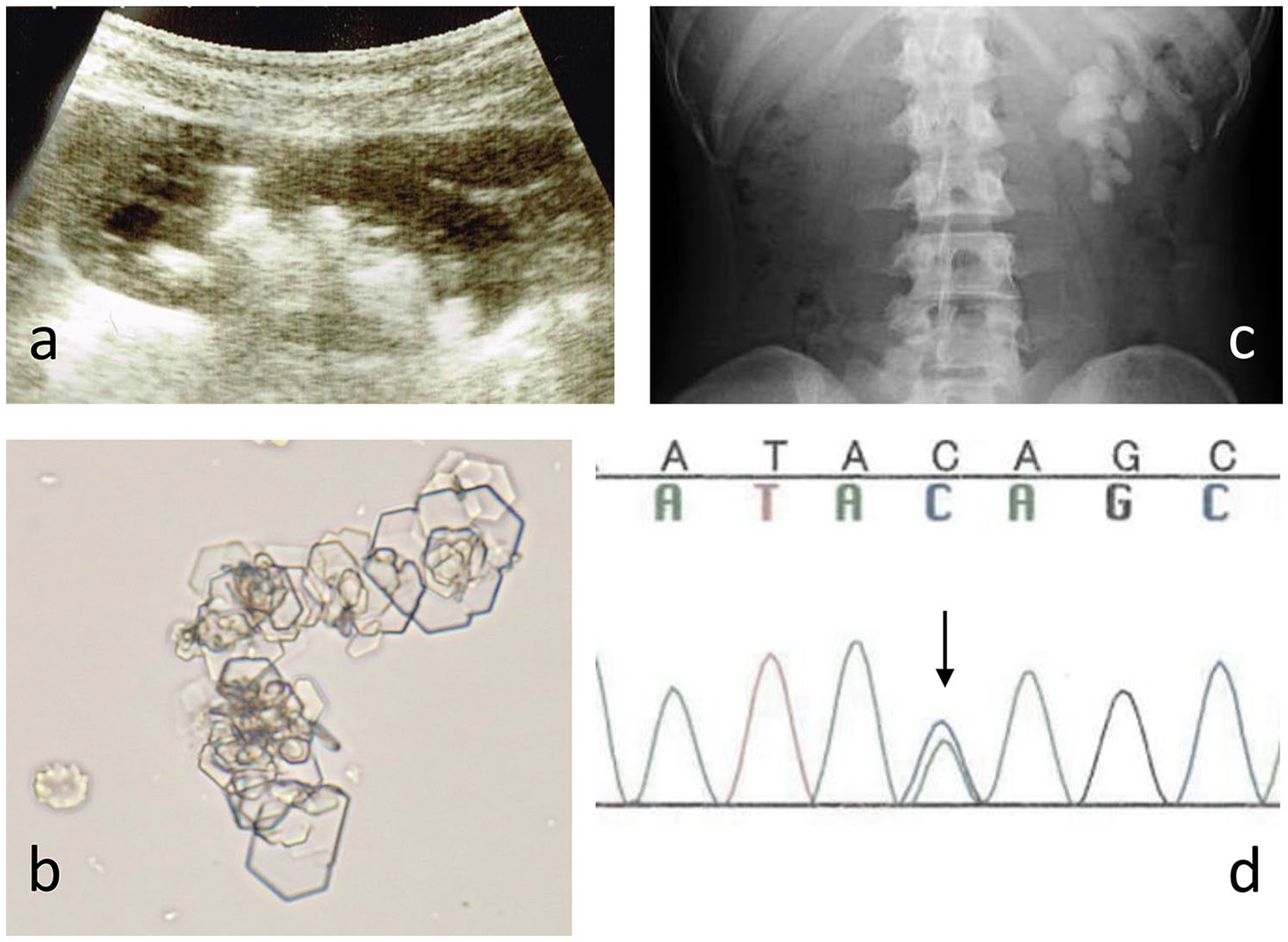
(a) Ultrasonography of the left kidney. Ultrasonography revealed hyperechoic lesions with sonic shadows, indicating urolithiasis. (b) Cystine crystals. The urinary sediments show hexagonal crystals, indicating cystine crystals. (c) Abdominal X-ray findings. Abdominal X-ray showed a staghorn calculus at the left pelviureteric junction. (d) DNA sequencing of SLC3A1. DNA sequencing showed a heterozygous mutation in SLC3A1. A nucleotide substitution c.1113C>A (Y371X) was detected (arrow).
At 9 years of age, we performed sequence analysis of SLC3A1 and SLC7A9 in the patient and her parents, from whom informed consent was obtained under ethical approval (Showa University, Human Genome, Gene Analysis Research Ethics Committee, No. 197). As shown in Figure 1d, the analysis revealed that she had a novel heterozygous mutation (c.1113C>A, Y371X). Her mother had no mutation. Her father had heterogeneous mutations in SLC3A1 (c.1113C>A, Y371X) and polymorphism of SLC7A9 (c.667>A, L233M) without cystinuria. His urinary concentrations of cystine, ornithine, lysine, and arginine were 82.1, 8.9, 247.6, and 11.7 µmol/g·Cr, respectively.
Discussion
The index case was a 4-year-old female patient with cystinuria that was discovered by chance. She had a novel heterozygous stop mutation in SLC3A1 (c.1113C>A), which was likely to be the cause of cystinuria.
Cystinuria is a global disorder with population-specific prevalence that varies between different populations. The highest frequency of cystinuria has been observed among Libyan Jews at a rate of 1:2500. Other population-specific rates are 1:17 000 in the United States, 1:18 000 in Japan, and 1:100 000 in Sweden.1-3 The mean age at which urolithiasis including cystinuria is diagnosed is reported to be 5.59 years. Of these patients, 41.4% were below the age of 1 year and 60.5% were below the age of 5 years. 4 Our patient was diagnosed at the age of 4 years, which is consistent with those reported. Symptoms at presentation are flank pain, restlessness, hematuria, urinary tract infections, and leukocyturia.4,5 Cystine stones are present in 6% to 8% of all pediatric nephrolithiasis patients. 1 The patient described here had hematuria and staghorn calculus but did not have flank pain or infection. Cystinuria was discovered by chance when investigating abnormal serum liver enzymes, while the cause of the elevated serum liver enzymes is unknown, and there are no previous reports on the association between cystinuria and elevated liver enzymes.
Although stones may be formed at any age, and more than 80% of patients develop their first stone within the first 2 decades of life, the first stone episode does not always occur in childhood. 6 Approximately 20% of cystinuria patients have staghorn calculi, with associated impaired renal function in 80% of these patients. 7 In this case, the patient had a staghorn calculus but did not have a stone episode. Akakura et al reported that the prognosis of urinary stone in Japanese patients with cystinuria is relatively good, and no patients in their study suffered from chronic renal failure. 8 However, it was recently reported that chronic kidney disease and high blood pressure often occur in patients with cystinuria. 9
Approximately 130 and 95 mutations have been reported in SLC3A1 and SLC7A9, respectively.10-12 The most common SLC3A1 mutation is R270X, which has been identified in 73% of Ashkenazi Jewish patients, and it accounts for 11% of the known mutations in North American patients. 1 The Y151C mutation is mainly observed in Northern European patients, 1 while the duplication dupE5E9 may have arisen in the German population. 1 The functional consequences of SLC7A9 mutations are generally broader than those of SLC3A1 mutations. 13 In a cohort of Japanese cystinuria patients, the c.1533C>T (P482L) mutation in SLC7A9 was present in over 80% of the cases, whereas SLC3A1 mutations were found in approximately 10%. 13 Reported mutations in SLC3A1 include deletion of T at nucleotide 1820 as well as heterozygote c.548T>C (V183A) and c.2017T>C (C673R) mutations. 13 The heterozygous Y371X mutation in SLC3A1 discovered in our patient has not been previously reported. Our patient had a novel heterozygous mutation in SLC3A1 (c.1113C>A), which changes the tyrosine at location 371 to a stop codon (Y371X), leading to insufficient translation of the protein and dysfunction of the transporter. It was unclear why the father was asymptomatic despite having the mutation in SLC3A1 (c.1113C>A, Y371X) and SLC7A9 (c.667>A, L233M). L233M is apparently a polymorphic change. 13 Urinary pH, gender, and/or extent of cystine reabsorption might be associated with the difference in phenotype. Furthermore, except the exon, additional mutation in the intron might affect the splicing of the mRNA that caused the defect in the protein synthesis in the patient. We are currently planning the next-generation sequence that may elucidate the mechanism further.
Eggermann et al proposed the following classification of cystinuria: Type AA, homozygosity for 1 SLC3A1 mutation or compound heterozygosity for 2 SLC3A1 mutations; Type A?, heterozygosity for 1 SLC3A1 mutation and a second unidentified mutation; Type BB, homozygosity for 1 SLC7A9 mutation or compound heterozygosity for 2 SLC7A9 mutations; Type B?, heterozygosity for a SLC7A9 mutation and a second unidentified mutation; Type AB, mixed heterozygosity of 1 SLC3A1 mutation and 1 SLC7A9 mutation; and Type AAA/AAB/ABB, 3 SLC3A1/SLC7A9 mutations in the same patient. 1 Hence, Type A? cystinuria is proposed as the diagnosis based on this classification.
Cystinuria is also observed in patients with cystinuria-hypotonia syndrome, which is due to a microdeletion of a part of SLC3A1 on chromosome 2q21. It is characterized by generalized hypotonia at birth, failure to thrive, growth retardation, and nephrolithiasis. 14 We consider that cystinuria in our patient was not associated with cystinuria-hypotonia syndrome, as our patient did not show any developmental delays.
In conclusion, a novel heterozygous point mutation that created stop codon, c.1113C>A (Y371X), was determined to be the cause of cystinuria in this case.
Footnotes
Author Contributions
YW: Contributed to analysis; drafted the manuscript; critically revised the manuscript gave final approval; agrees to be accountable for all aspects of work ensuring integrity.
YA: Contributed to conception and design; contributed to analysis; drafted the manuscript; critically revised the manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity.
SS: Contributed to conception and design; contributed to gene analysis; critically revised the manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity.
EM: contributed to urine analysis; gave final approval.
YT: contributed to analysis; gave final approval.
SH: contributed to analysis; gave final approval.
TF: contributed to analysis; gave final approval.
SW: contributed to analysis; gave final approval.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: A part of this work was supported by 5th Grant Aid from the Japanese Society on Urolithiasis Research.