
Abstract
RASopathy is caused by dysfunction in the MAPK pathway, and include syndromes like Noonan syndrome (NS), NS with multiple lentigines (formerly known as Leopard syndrome), cardiofaciocutaneous (CFC), Legius syndrome, capillary malformation–arteriovenous malformation, neurofibromatosis type 1, and Costello syndrome. When counted together, RASopathies affect 1/1000 live births, and are characterized by cardiovascular manifestations, short stature, developmental delay, renal, urogenital, skin/skeletal abnormalities, and dysmorphic appearance. NS—one of the most common RASopathies—occurs in 1/1000 to 1/2500 live births. On the other hand, the frequency of CFC is unknown, but it is one of the rarest RASopathies, with estimates of only a few hundred cases worldwide. However, its phenotype overlaps with that of NS. In this case series, we describe 5 patients with a clinical and genetic diagnosis of RASopathy—either NS or CFC—all of whom were also diagnosed with isolated sagittal synostosis (ISS). Medical records from ophthalmology, cardiology, plastic surgery, medical genetics, cleft craniofacial, and neurosurgery were used to determine patient history. In our cohort, late presentation of ISS was the predominant form of ISS presentation. We hope this report further characterizes the burgeoning relationship between RASopathy and ISS. Furthermore, these findings support including sagittal synostosis among the presenting features in the clinical phenotype of RASopathies. Ethical approval was obtained from the university’s institutional review board.
Methods
Retrospective review of patients diagnosed with isolated sagittal synostosis (ISS). Patients were identified through perusal of clinical databases at a single institution. Ethical approval was obtained from the University of Pittsburgh Institutional Review Board (IRB), Protocol PRO16110394. The study was granted a waiver to obtain informed consent and a waiver to the requirement of written Health Insurance Portability and Accountability Act authorization to collect protected health information by the IRB due to the minimal risk involved in the study, the fact that the rights of the patients would not be violated by collecting this information, and that this research could not have been conducted without these waivers. Decision for surgery was determined by the craniofacial team, based on the patient’s craniocephalic index, overall deformity, as well as risk for functional and developmental deficits and/or raised intracranial pressure.
Background
RASopathy is caused by dysfunction in the MAPK pathway, resulting in a group of diseases including Noonan syndrome (NS), Costello syndrome (CS), and cardiofaciocutaneous (CFC) syndrome. RASopathy may result in congenital and or acquired cardiomyopathy, short stature, developmental delay, renal, urogenital, skin/skeletal abnormalities, and dysmorphic appearance. The mechanism of Ras dysfunction begins with the disruption of the intrinsic GTPase activity of the Ras protein, which causes excessive Ras signaling. This may lead to exaggerated cell growth and even cancer. 1 The aim of this article is to further characterize the association between ISS and RASopathy, as demonstrated in 5 patients seen at a single institution. IRB approval was obtained before acquisition of patient information. Informed consent was obtained for the use of patient photographs.
Patient 1 (KRAS)
This male was born at 37 weeks and diagnosed with cryptorchidism 1 month after birth. He presented with macrocephaly, gastroesophageal reflux disease, prominent pectus, holosystolic murmur, dysmorphic facies, bilateral arachnoid cysts, developmental delay, hypotonia, and submucous cleft palate. He underwent computed tomography scan at 5 months due to macrocephaly, confirming ISS. The patient was diagnosed with NS, molecularly confirmed by a mutation in c.40G>A (p.Val14Ile) of the KRAS gene. Ophthalmic examination revealed amblyopia and nystagmus oculus uterque (both eyes). Fundoscopy revealed optic nerve hypoplasia, optic nerve pallor, and papilledema oculus uterque. The patient underwent craniectomy and barrel stave osteotomy to reduce intracranial pressure at 2.1 years of age.
Patient 2 (BRAF)
This female born at 38 weeks presented with moderate patent ductus arteriosus, patent foramen ovale, and enlarged liver shortly after birth. By 17 months, developmental delay and hypotonia were observed. Magnetic resonance imaging determined nonspecific cortical and central white matter volume loss with corpus callosal thinning. Genetic testing revealed a mutation of c.1741A>G (p.Asn581Asp) in the BRAF gene, confirming a diagnosis of CFC. At 4 years, her scaphocephaly was noted, and she underwent a computed tomography scan, confirming a diagnosis of ISS at 4.5 years of age. She did not undergo surgical repair (see Figures 1 and 2).

Three-dimensional computed tomography scan (patient 2) demonstrating coronal, axial, and sagittal views of patient with isolated sagittal synostosis and associated scaphocephaly.
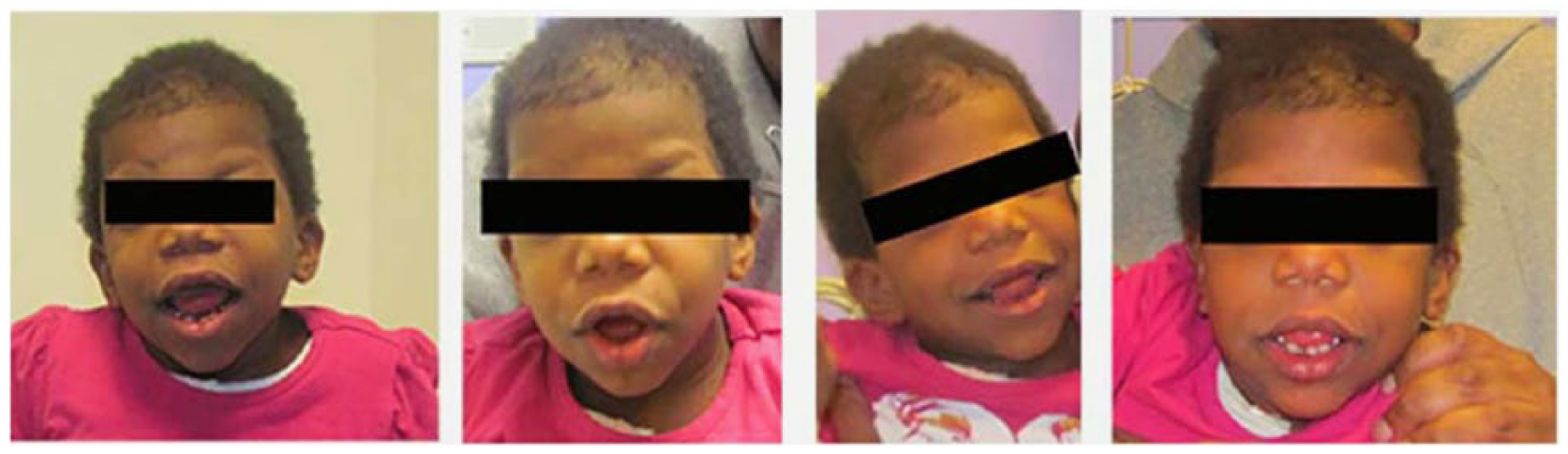
Photos (patient 2) demonstrating isolated sagittal synostosis and RASopathy.
Patient 3 (BRAF)
This 36-week premature male was seen at 2.5 months for heart murmur and was diagnosed with mild pulmonary stenosis (PS) and atrial septal defect (ASD). By 6 months, his developmental delay and poor weight gain were noted. He was diagnosed with idiopathic neutropenia, hypertonia, right inguinal hernia, and autism. Magnetic resonance imaging identified periventricular white matter loss and thinning of the corpus callosum. He was scaphocephalic and diagnosed with ISS at 2.4 years of age. Afterwards ophthalmology noted optic nerve atrophy, pallor, Brown syndrome, and abnormal visual evoked potential testing results. Exome sequencing revealed a de novo BRAF mutation: heterozygous c.1497A>C, (p.K499N), confirming his diagnosis of CFC. This patient suffers from severe speech delay.
Patient 4 (BRAF)
This 34-week premature male appeared scaphocephalic at birth and presented with failure to thrive, ASD, PS, and pyloric stenosis. He underwent surgery for closure of his ASD and for release of PS. The patient was diagnosed with ISS at 5 months of age and underwent surgical repair at 1.5 years of age via right and left frontal craniectomies. His ophthalmic examinations revealed normal physiology but abnormal visual evoked potential testing. This patient has significant developmental and speech delays. Medical genetics suspected NS, which was confirmed when genetic testing identified a c.735A>T (p.Leu245Phe) mutation in the BRAF gene.
Patient 5 (PTPN)
This male born at 37 weeks demonstrated significant PS and slightly dysmorphic features. He was noted to have a horseshoe kidney, developmental delay, short stature, staring spells, language delay, and submucous cleft palate. His scaphocephaly was noted at 3 years, and he was diagnosed with ISS and right positional plagiocephaly at 3.5 years. He did not undergo surgical repair for ISS. Telecanthus and epicanthus were noted at ophthalmological examination. His clinical diagnosis of NS was confirmed via genetic testing, which identified the following mutation in PTPN11 gene: c.188A>G, (p.Tyr63Cys).
Discussion
We identified 1 patient with a KRAS mutation associated with NS, 2 patients with BRAF mutations associated with CFC, 1 patient with a BRAF mutation associated with NS, and 1 patient with a PTPN mutation associated with NS. Each of these patients presented with traits characteristic of NS or CFC and was diagnosed with ISS (no other sutural involvement). There is increasing evidence suggestive of a relationship between ISS and RASopathy. At least 6 case reports describing craniosynostosis in patients with RASopathy—namely NS, CS, and CFC—have been published since 2009.2,4-8 CS patients with RASopathy have predominantly had scaphocephaly, even though multi-sutural and pan-sutural involvement have been reported.2,4,7,8 Mutations in the following genes have been identified in patients diagnosed with RASopathy and craniosynostosis: SHOC2, KRAS, BRAF, and PTPN1. We have identified patients with mutations in the KRAS, BRAF, and PTPN11 genes. Though KRAS mutations are believed to account for fewer than 5% of genetic diagnoses of NS, 6 our cohort—as well as all but one case report describing scaphocephaly with RASopathy—include at least 1 patient with a KRAS mutation.2,3,5,6 This cohort of patients with ISS and RASopathy demonstrates a strong pattern in pathology (see Table 1). Developmental and speech delay are found in 5/5 patients. The relationship between RASopathy and speech delay, especially in patients with CFC, has been previously described. 9 Furthermore, the relationship between CS and abnormal speech/language development has been estimated to be as high as 1 in 1.7 patients with nonsyndromic craniosynostosis. 10 The authors determined that more than half of nonsyndromic CS patients have abnormal speech and language development, and that 1/3.4 patients deficits were sufficient to warrant speech therapy. In our cohort, 4/5 patients (80%) diagnosed with RASopathy and ISS have late onset ISS. Previous research has not distinguished between early- and late-onset synostosis in patients diagnosed with RASopathy and CS. We believe age of onset to be a key variable in ISS presentation and treatment. Furthermore, the predominance of late-onset ISS in patients diagnosed with both ISS and RASopathy warrants long-term monitoring by a craniofacial specialist for late-onset ISS patients. Patients like those described here may not present with typical sequelae of ISS or RASopathy at their preliminary evaluation, but may develop illness later in life. Continued monitoring by medical genetics may also be crucial, considering 40% of patients with CFC do not yet have molecular confirmation of their diagnosis. 11 Hopefully, through increased genetic testing of patients with RASopathy and ISS, clinicians will be able to offer an accurate genetic diagnosis of RASopathy to a greater proportion of patients and their families.
Pathology in patients with ISS and RASopathy.

Abbreviations: ISS, isolated sagittal synostosis; CT, computed tomography; CFC, cardiofaciocutaneous; PS, pulmonary stenosis; PDA, patent ductus arteriosus; PFO, patent foramen ovale; ASD, atrial septal defect; N/A, not applicable.
Conclusion
Here we describe the largest cohort of patients with RASopathy and sagittal synostosis from a Western population. Late-onset synostosis predominates in our cohort, which warrants long-term monitoring of patients with ISS and/or RASopathy. Our research is the first to suggest strong patterns in developmental pathology that may be expected in patients diagnosed with RASopathy and ISS as they grow older. For example, all patients in our cohort suffered from speech delay. Regarding treatment and care, we believe ISS should be among the list of presenting traits in patients with RASopathy. The mutations observed in our cohort further substantiate the need for expanding the clinical features of NS to include craniosynostosis. The clear pattern of pathology observed in these patients demonstrates a need for long-term monitoring, speech therapy, and other care. Finally, it calls for increased genetic testing of patients with sagittal synostosis, as this will increase our knowledge of genetic loci underlying the clinical presentation of these children.
Footnotes
Author Contributions
AD: has examined patient records and identified patients who had been previously diagnosed with ISS, drafted and revised the manuscript accordingly.
GZ: confirmed each diagnosis of Craniosynostosis identified via radiologic review
MH: examined patient records and identified patients who had been previously diagnosed with ISS, drafted and revised the manuscript accordingly
JL: Author of patient records and attending surgeon to the study’s patients
IP: Editor, and author of patient records and attending surgeon to the study’s patients
JG: Editor and author of patient records and attending surgeon to the study’s patients
SM:Khetarpal: Editor and author of patient records and attending surgeon to the study’s patients
KN: Principal Investigator and corresponding author
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.