
Abstract
Objective: To pilot a newborn screening program for sickle cell disease (SCD) in St. Vincent and the Grenadines using a novel partnership method to determine the feasibility of a universal newborn screening program in this country. Methods: A prospective study of mothers and their newborns was conducted between January 1, 2015, and November 1, 2015, at the country’s main hospital. Mothers of infants born at this hospital were offered screening for SCD for their infants. If accepted, the newborn’s heel-stick blood specimen was obtained and mailed to the South Carolina Department of Health and Environmental Control Newborn Screening Laboratory for testing. Samples were analyzed for variant hemoglobins using standard laboratory techniques and results were communicated to local physicians. Feasibility was determined by a benchmark of having >50% of SCD patients receive the diagnosis and initiate disease-specific care by 3 months of age. Descriptive statistics were completed using SAS 9.4. Results: There were 1147 newborn infants screened for SCD. Of these, 123 (10.7%) had results indicative of sickle trait and 3 patients (0.3%) were diagnosed with SCD: 1 with HbSS and 2 with HbSC. All 3 patients with SCD received treatment before 3 months of age. Conclusions: A newborn screening program is feasible in this population when partnered with an established newborn screening laboratory.
Introduction
Sickle cell disease (SCD) results from a qualitative defect in β-hemoglobin chain synthesis and is one of the most common genetic disorders worldwide. 1 Individuals affected by this disease experience reduced quality of life and have a shortened life span as a result of the multi-organ injury that occurs. 2 Newborn screening for SCD, when coupled with comprehensive care, has been shown to decrease the early mortality associated with the disease.3,4
In the United States, where there is universal newborn screening for SCD, the disease is seen in approximately 1:375 African American births. 5 Data from the Caribbean show higher incidence rates among all newborns. In Guadeloupe, the incidence of SCD is 1:304 newborns, and the rate is 1:150 newborns in Jamaica.6 -8 Despite the higher burden of disease in the Caribbean, most countries in this region do not have newborn screening programs for SCD. Jamaica introduced limited newborn screening in 1995. 6 Following the initiation of that program, a Jamaican study showed that the under 5 years of age mortality rate for children with SCD was the same as the general population when those children were diagnosed by newborn screening and given disease-specific care. 9 Similarly, studies in other resource limited settings have shown that newborn screening for SCD can be cost-effective due to the prevention of complications.10,11 Despite the demonstrated effectiveness of newborn screening for SCD, many resource-limited nations have not been able to mobilize the necessary infrastructure or expertise to initiate this type of program.
St. Vincent and the Grenadines (SVG) is a multi-island nation in the Caribbean with a total population of around 109 000 and 1700 to 1800 births per year. 12 There are no newborn screening programs in this country, and the incidence of SCD in this population is largely unknown. Our recent retrospective study estimated that the prevalence rate among live births was 1:172, and that most patients are diagnosed at more than 1 year of age. 13 As there are no newborn screening programs, most patients are identified when they present with symptoms and the diagnosis confirmed by qualitative electrophoresis, which is available at the country’s main hospital. Once the diagnosis is confirmed, prophylactic penicillin is initiated and children are followed in local public clinics or in private physician’s offices as there are no specialty clinics for SCD in SVG. The penicillin is usually given as monthly intramuscular injections, which is administered at either a local public clinic or a private physician’s office.
The objective of this study was to assess the feasibility of a newborn screening program for SCD in SVG through a novel partnership approach between the major birthing hospital in SVG and an established newborn screening laboratory in the United States. Our goal was to assess this partnership screening approach in a country where pediatric care and prophylactic penicillin are available, but whose size and population limit the establishment of an independent newborn screening laboratory. We defined feasibility by the benchmark of having >50% of SCD patients receive the diagnosis and initiate disease-specific care before 3 months of age.
Methods
This study was approved by the Institutional Review Board of the Medical University of South Carolina and the National Ethics Review Committee of the SVG Ministry of Health, Wellness and the Environment. Although the study had the support and approval of the SVG Ministry of Health, this was not a government-mandated public health initiative and was therefore conducted as human subjects research. The local SVG research team underwent human subjects research training and certification prior to initiation of this study, and informed consent was required for every study participant.
The study was conducted at the Milton Cato Memorial Hospital (MCMH), which accounts for about 95% of all newborn deliveries in SVG and is the only secondary care referral hospital in the nation where specialized care can be obtained. 12
The study population were newborns who were delivered between January 1, 2015, and November 1, 2015. Prior to hospital discharge, mothers were approached and informed about participation in a study of newborn screening for SCD. Prescreening education was performed by our local research team, and pamphlets were given to the mothers that provided information on SCD, sickle trait, and the newborn screening process. If participation was accepted, a signed consent was obtained and the infant’s specimen was collected. Both well newborns and infants admitted to the neonatal intensive care unit were included and there were no study exclusion criteria. For infants admitted to the neonatal intensive care unit, study protocol ensured that maternal consent and specimen collection occurred prior to the transfusions of any blood products.
Prior to the initiation of the study, the research team and the house staff at MCMH were trained on specimen collection techniques. The newborn screen specimen was obtained via a heel stick and the blood was impregnated on standard specimen collection forms obtained from the South Carolina Department of Health and Environmental Control Newborn Screening Laboratory (SC DHEC NBS Lab). Specimen forms were left to dry for at least 4 hours and then kept at room temperature until they were sent in batches every 2 weeks, via air courier to the SC DHEC NBS Lab. Previous studies have shown that samples can be stored at room temperature for weeks and still allow for accurate hemoglobin identification using current screening techniques. 14 The SC DHEC NBS Lab performs all newborn screen blood tests for the state of SC.
Once received by the laboratory in South Carolina, testing was performed using standard laboratory operating procedures. The current screening algorithm is to first test all samples using isoelectric focusing. Specimens that show Hb variants are then retested using high performance liquid chromatography (HPLC), which is able to quantify the abnormal Hb variants found. All results were then mailed from the SC DHEC NBS Lab to the physician team in SC. Any results that were indicative of SCD were also faxed (on the day they were resulted) to the SC physician team to ensure timely follow-up.
Once results were reviewed by the SC physician team, the results were called to the SVG local team and entered into the secure research database. All results were reviewed a second time to ensure accuracy. The database results were accessible to study physicians both in SC and SVG. Parents were then contacted by the SVG local team and patients with abnormal results were given appointments to the central pediatric clinic located at the study site (MCMH). During these visits, families of infants diagnosed with SCD received counselling on the disease and disease-specific care was initiated based on the current local SCD standard of care. Families of infants diagnosed with sickle trait and other variant hemoglobins also received posttest counselling and any infant requiring local confirmatory testing in the form of Hb electrophoresis was arranged as needed (Figure 1).

Process map of study methods.
For all infants enrolled in the study, we collected demographic information, maternal sickle cell status, family history of SCD, and the infant’s follow-up physician or follow-up clinic at discharge. Study data were collected and managed using REDCap (Research Electronic Data Capture) electronic data tools hosted at the Medical University of South Carolina. REDCap is a secure web-based database application designed to support data capture for research studies. 15 Descriptive analyses were performed using SAS9.4 (SAS Institute, Cary, NC). Geographical data were analyzed and displayed using the geographic information system QGIS 2.18.0. 16
Feasibility was assessed by the benchmark of having >50% of SCD patients receive the diagnosis and initiate disease-specific care before 3 months of age.
Results
During the study period, 1147 newborns underwent screening for SCD. Newborns represented all parishes in the mainland of St. Vincent and a number of children were also from the Grenadine islands (Figure 2). A total of 586 (51.1%) of newborns were males and 561 (48.9%) were female. There were 14.3% mothers who reported a positive family history of SCD and 8.5% of infants were born to mothers who were known carriers of abnormal hemoglobin variants.
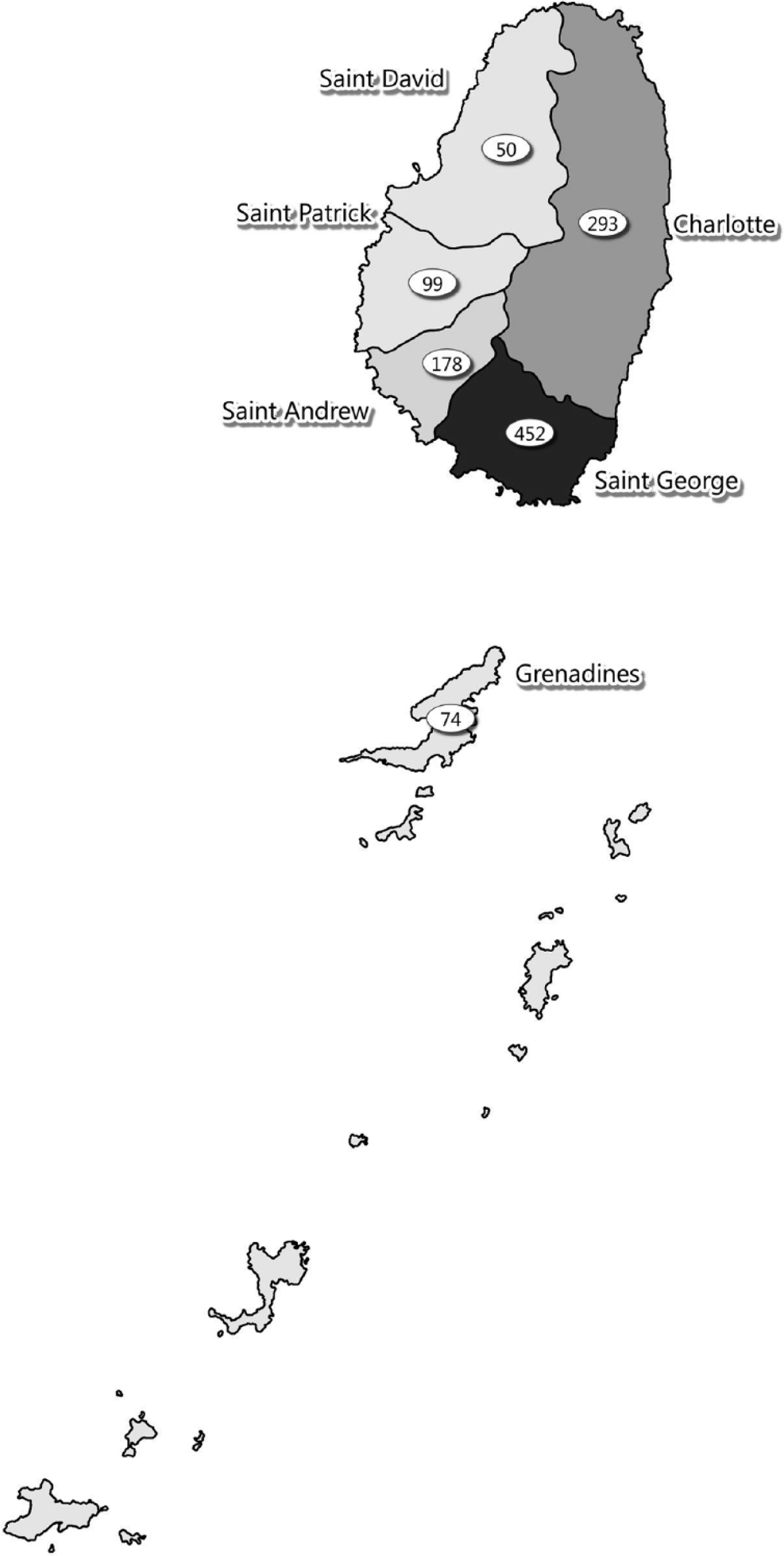
Distribution of newborns in study population by parish.
The median age at testing was 1 day of age. There were no unsatisfactory specimens as determined by the SC DHEC NBS Lab; therefore, there were no requests for repeat samples. All heel stick specimens were successfully received by the laboratory and all results were successfully received by SVG physicians.
There were 165 (14.4%) infants with abnormal hemoglobin variants on HPLC testing (Figure 3). Of these, 123 (10.7%) had results indicative of sickle trait, and 3 patients (0.3%) had results indicative of SCD: 1 with HbSS and 2 with HbSC. Hemoglobin C trait was seen in 27 (2.4%) of infants (Table 1).
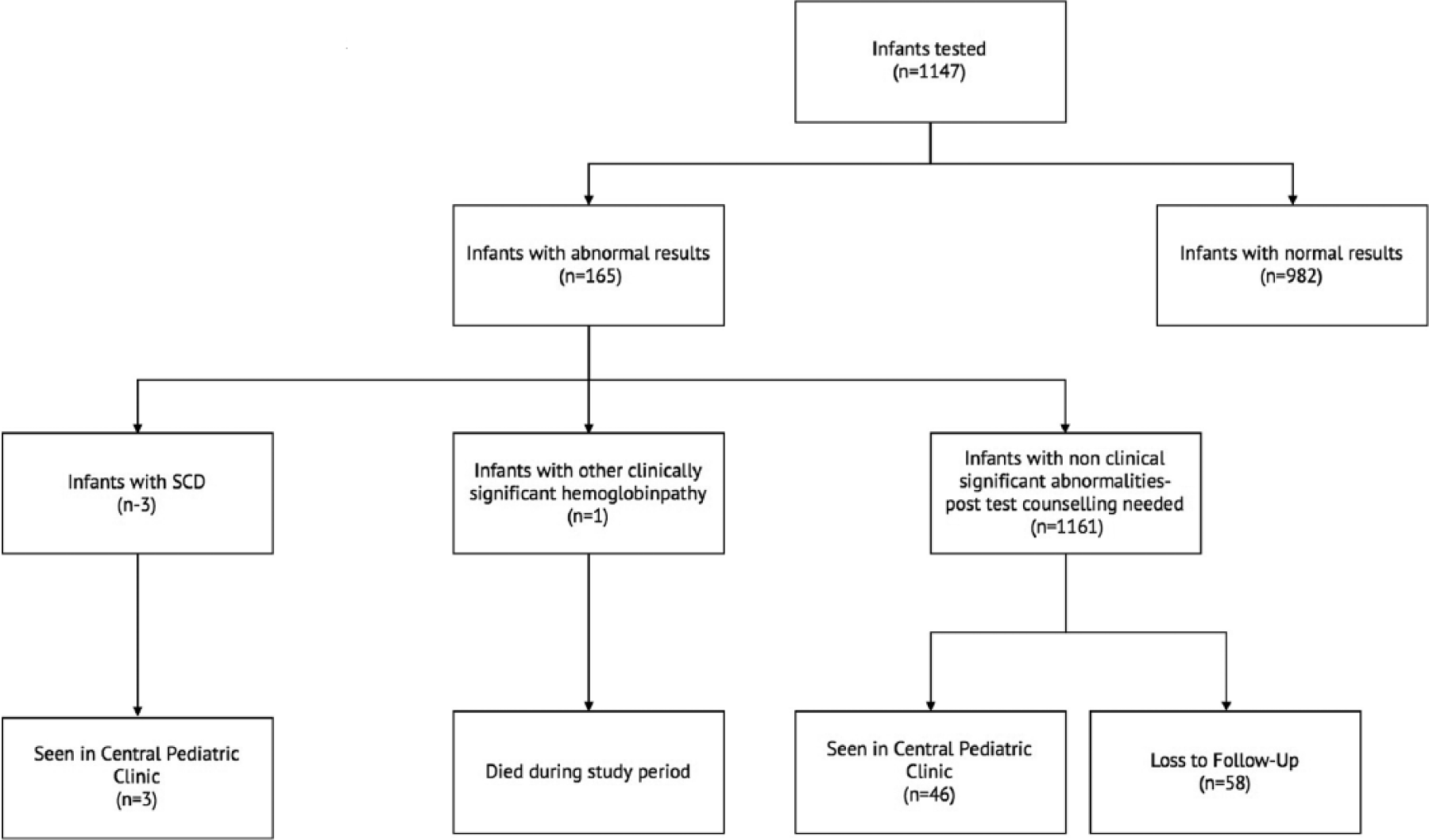
Flow diagram of study population.
Distribution of Newborn Screen Results in the Study Population (N = 1147).

Abbreviations: HPLC, high-performance liquid chromatography; SCD, sickle cell disease.
Of the infants with abnormal hemoglobin variants identified, 105 (64.8%, N = 164) were followed-up in the central pediatric outpatient clinic for discussion of their results (Figure 3). All 3 infants who were diagnosed with SCD were seen in the clinic in the first 2 months of age. The median time to discussion of results for all patients was 3 months (range = 0-10 months).
There was one neonatal death during the study period. This infant passed away a few days after birth from complications related to prematurity and severe growth restriction. The newborn screen results were not yet available at the time of death, but were later identified as FF, suggesting β-thalassemia major.
Our benchmark for feasibility was determined if more than 50% of infants with SCD received a diagnosis and establish disease-specific care by 3 months of age. In our study, 100% of infants diagnosed with SCD received a diagnosis that was communicated to the family and established disease-specific care by 3 months of age.
Discussion
Our study demonstrated that this partnership approach to newborn screening for SCD was feasible for St. Vincent and the Grenadines and could likely be used in other underresourced areas. All 3 infants with SCD were diagnosed and began disease-specific care in a timely manner (within the first 2 months of age), which is in keeping with the standard of care as outlined by the American Academy of Pediatrics. 17 This result was higher than our benchmark for feasibility, which was set as >50% of infants receiving diagnosis and establishing disease specific care by 3 months of age.
Likewise impressive, there were no unsatisfactory or indeterminate blood spot specimens. All specimen collection was performed by local staff who had never used these techniques prior to the training they received during this study. This testing success is an important accomplishment as reports have documented rates of unsatisfactory specimens as high as 7.7% for some states in the United States. 18 These findings further suggest the feasibility of this approach for SVG.
Numbers from the United States show that SCD affects around 1:1900 newborns and sickle trait is seen in 1:67 newborns nationally. 19 In our cohort of infants born from January 1, 2015, to November 1, 2015, SCD was found in 1:382 births and the sickle trait was seen in 1:9. Our previous retrospective study of SCD birth rates estimated 1:112 for birth year 2006 and 1:364 for birth year 2007. Our current study rates for SCD of 1:382 approximates our previous estimates for 2007.
Using data obtained from the MCMH delivery records, our local SVG study physicians determined that there were 1371 live births at MCMH during our study period (personal communication). Although we did not collect the number of refusals to participate in this study, nor the number of mothers “missed” for invitation to this study, we estimate that 83.6% (1147/1371) infants born at the MCMH during the study period accepted and received testing.
In SVG, the current practice for SCD screening is to screen all pregnant females without a known history of SCD for sickle trait using the sodium metabisulfite test. This test is referred to as the “sickle screen.” It is a qualitative visual detection test that cannot distinguish between SCD or sickle trait nor can it be used to identify other abnormalities of the β-hemoglobin gene. 20 Since this test requires a minimum of 10% HbS, it cannot be performed on newborns. 20 Infants born to mothers who have been found to have the sickle trait on prenatal testing are then offered screening for their infants at 6 months of age. At 6 months of age, these infants are tested using this “sickle screen” and those with positive results undergo confirmatory testing with Hb electrophoresis. This method offers a targeted infant screening approach that relies only on the maternal sickle cell status as determined by an unreliable primary screening test. At best, this approach may identify some affected infants at 6 months of age. However, in our previous study the earliest age at diagnosis was at 8 months and most patients were diagnosed at over the age of 12 months. 13 These results suggested that this targeted approach leads to delays in diagnosis and that patients are more likely to present symptomatically. 13
Of the 3 infants who were diagnosed with SCD during this pilot project, only one had a mother who was a known carrier of an abnormal hemoglobin variant (sickle trait). In the other 2 cases, the mother’s sickle cell status at delivery was documented as negative. Therefore, these 2 infants would not have been referred for screening according to the current local screening practice at 6 months and would likely not have been diagnosed until they became symptomatic. Thus, the current practice of targeted infant screening is not an alternative to newborn screening, which allows for diagnosis and initiation of preventative therapies prior to the onset of symptoms.
The world has become a global village and we continue to see how partnerships between nations can be the bridge to improving the health care in lower and middle income countries. 21 As seen in our study, a partnership with an established newborn screening laboratory can be a feasible way to develop newborn screening in countries where the need exists, but where the infrastructure might not allow for the establishment of an in-country laboratory. Our results suggest that this partnership newborn screening program for SCD is feasible and valuable in SVG.
Author Contributions
SAW: Contributed to conception and design; contributed to acquisition, analysis and interpretation; drafted the manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
BBF: Contributed to conception and design; contributed to acquisition; critically revised the manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
YS: Contributed to acquisition; critically revised the manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
KM: Contributed to analysis; critically revised the manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
SGR: Contributed to conception and design; critically revised the manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
JK: Contributed to conception and design; critically revised the manuscript; gave final approval; agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Footnotes
Acknowledgements
We would like to thank the South Carolina Department of Health and Environmental Control Newborn Screening Laboratory and the Ministry of Health, Wellness and the Environment of SVG.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by support in part by the faculty pilot grant offered through the Center for Global Health, Medical University of South Carolina, Charleston, SC, with NIH/National Center for Research Resources Institutes Award Number UL1TR000062. Additional support was received though the South Carolina Clinical &Translational Research (SCTR) Institute, with an academic home at the Medical University of South Carolina, through NIH Grant Numbers UL1RR029882 and UL1TR000062.