
Abstract
Acoustic shocks and traumas sometimes result in a cluster of debilitating symptoms, including tinnitus, hyperacusis, ear fullness and tension, dizziness, and pain in and outside the ear. The mechanisms underlying this large variety of symptoms remain elusive. In this article, we elaborate on the hypothesis that the tensor tympani muscle (TTM), the trigeminal nerve (TGN), and the trigeminal cervical complex (TCC) play a central role in generating these symptoms. We argue that TTM overuse (due to the acoustic shock), TTM overload (due to muscle tension), and ultimately, TTM injury (due to hypoxia and “energy crisis”) lead to inflammation, thereby activating the TGN, TCC, and cortex. The TCC is a crossroad structure integrating sensory inputs coming from the head–neck complex (including the middle ear) and projecting back to it. The multimodal integration of the TCC may then account for referred pain outside the ear when the middle ear is inflamed and activates the TGN. We believe that our model proposes a synthetic and explanatory framework to explain the phenomena occurring postacoustic shock and potentially also after other nonauditory causes. Indeed, due to the bidirectional properties of the TCC, musculoskeletal disorders in the region of the head–neck complex, including neck injury due to whiplash or temporomandibular disorders, may impact the middle ear, thereby leading to otic symptoms. This previously unavailable model type is experimentally testable and must be taken as a starting point for identifying the mechanisms responsible for this particular subtype of tinnitus and its associated symptoms.
Introduction: Symptom Cluster Resulting From Acoustic Shock
An acoustic shock is defined as a sudden, unexpected advent of a brief, moderately loud to loud sound stimulation (McFerran & Baguley, 2007; Milhinch, 2001; Westcott, 2006). Although the duration and level of these sound stimulations are rarely sufficient to result in permanent cochlear damage, they can, in certain cases, produce several varying and troublesome symptoms such as tinnitus, hyperacusis, ear fullness (feeling of abnormal pressure in the middle ear) and ear tension, dizziness, vertigo, and pain in and outside the ear. These symptoms are most often temporary and disappear within a few hours or days following the acoustic shock. However, in some cases, they can become chronic and seriously impact quality of life (Patuzzi, Milhinch, & Doyle, 2002; Westcott, 2016; Westcott et al., 2013).
It has been suggested that the tensor tympani muscle (TTM) could play a central role in the onset of these symptoms (Westcott, 2006, 2016; Westcott et al., 2013). This hypothesis was inspired by the pioneering work of Klockhoff who suggested that the tonic tensor tympani syndrome (TTTS) could account for a similar symptom cluster than that reported after acoustic shock (Klockhoff, 1961, 1979; Klockhoff & Anderson, 1960). Klockhoff suggested that spontaneous TTM activity was responsible for fluctuation in the middle ear impedance called “the tonic tensor phenomenon.” Interestingly, among subjects showing the “tonic tensor phenomenon,” 83% reported ear fullness, 62% tinnitus, 42% dysacusis, 88% tension headache, and 80% dizziness and disequilibrium. Klockhoff (1979) further suggested that TTTS might be caused by “increased psychic tension due to mental stress” (pp. 69–76).
It is noteworthy to mention the prevalence of acoustic shock injuries, which is estimated to affect nearly 13% of call center workers (John, Poonamjeet, Peter, & Musa, 2015). While acoustic shocks have drastically brought into the light the potential role of the TTM in the onset of these symptoms, it is most likely that the same symptoms could result from other noise-hazard situations, such as occupational and leisure noise exposure (Stucken & Hong, 2014). Therefore, the occurrence of TTTS is likely more frequent than suspected. In this respect, a recent study tried to estimate the prevalence of TTTS in tinnitus and hyperacusis subjects by asking them to identify, within a list of symptoms potentially associated with TTM dysfunction, which symptoms they were affected by. In nonsevere hyperacusis subjects (with or without tinnitus), 81.1% reported at least one TTTS associated symptom and this percentage rose to 91.3% in severe cases. Interestingly, the feeling of ear fullness was the most reported symptom among all (52.6% of cases). For tinnitus subjects (without hyperacusis), the reported percentages were 40.6% and 68% for nonsevere and severe cases, respectively (Westcott et al., 2013). In conclusion, this study suggests that TTTS would be present in a relatively large proportion of tinnitus and hyperacusis subjects.
Although the hypothesis of TTM involvement in these symptoms was proposed almost 40 years ago (Klockhoff, 1979), the detailed mechanisms leading to TTTS remain elusive. In particular, it is unclear if the TTM is involved in otalgia and symptoms possibly resulting from the inner ear and central nervous system or how it could contribute to the apparition of these symptoms. The challenge undertaken in this article is to propose a potential premise explaining the emergence of these symptoms following an acoustic shock or trauma. This model may also provide a framework to account for the symptom cluster potentially related to nonacoustic events such as musculoskeletal disorders affecting the neck and head region (cervical and temporomandibular disorders—TMDs, head and neck trauma, or whiplash, for instance).
A Brief Overview of the Middle Ear
Evolutionary History of the Middle Ear
The middle ear emerged as vertebrates became terrestrial. The evolutionary history of the middle ear has puzzled morphologists for many years as the number of ossicles in mammals is different than that in amphibians, reptiles, and birds (Takechi & Kuratani, 2010). This fact was problematic, as it is believed that evolution does not usually result in the formation of new anatomic elements de novo. In other words, it is thought that every animal skeletal element is derived from changes to a common skeletal background. The challenge for morphologists was therefore to identify the homologs of mammalian ossicles in the nonmammalian skull.
The ossicles originate from the first two pharyngeal arches, the bony structures originally supporting the gills. The stapes (or columella in nonmammalian tetrapods) comes from the second pharyngeal arch and is homologous to the hyomandibular bone in fish, which plays a role in anchoring the mandibular arch to the skull bone (as of tetrapods; Anthwal, Joshi, & Tucker, 2013; Anthwal & Thompson, 2016). The malleus and the incus originate from the first pharyngeal arch. The first pharyngeal arch became structures allowing for jaw articulation in reptiles and birds, namely, articular and quadrate bones. Then, the quadrate and the articular bones have lost their function in the mammalian temporomandibular joint (TMJ), and these bones have become middle ear ossicles, namely, the incus and the malleus, respectively.
The evolutionary origin of the middle ear shows a functional proximity between the ossicles and the jaw articulation. Interestingly, the TTM and the TMJ muscles have the same innervation (mandibular branch of the trigeminal nerve). Based on the phylogenetic proximity of the TTM and jaw muscles, TTM has even been considered as a manducatory apparatus muscle (Ramirez, Ballesteros, & Sandoval, 2008). The close relationships between the TMJ and the TTM may have important functional consequences. In particular, TMJ disorders (TMJDs) may impact the TTM (see later).
Anatomy and Function of the Middle Ear
The mammalian middle ear consists of the tympanic membrane and three ossicles: the malleus (attached to the tympanic membrane), the incus, and the stapes (which is in contact with the oval window). It serves the purpose of adapting impedance between the air and the liquid that fills the inner ear, essentially through a surface difference between the tympanum and the oval window.
The eardrum is mostly innervated by the mandibular branch of the TGN but also by the facial, the vagus, and the glossopharyngeal nerves (Saunders & Weider, 1985; Uddman, Grunditz, Larsson, & Sundler, 1988). The middle ear mucosa is predominantly innervated by the mandibular branch of the TGN and in lesser extent by the maxillary branch of the TGN (Oyagi, Ito, & Honjo, 1990).
The middle ear is filled with an inert medium (air) that minimizes frictional forces, allowing the proper function of the middle ear complex. The middle ear is covered by a protective respiratory epithelium (mucosa) composed of ciliated and secretory cells including goblet cells and is richly vascularized (Ars et al., 1997; Hentzer, 1984). The mucosa can create folds at the entrance of the round window niche and can even partly cover it. The middle ear mucosa plays a double role: It is a defense system against infectious agents and is involved in the isobaric system of the middle ear through gas exchange. The mucosa cells have a limited life span and are constantly replaced by dividing and differentiating cells into new mature cells. A “problem” faced by the middle ear is therefore avoiding the accumulation of “mucosa wastes” in the middle ear cavity. One role of the many roles played by the Eustachian tube is precisely to eliminate these wastes. If the Eustachian tube does not function properly or if it is blocked or if the mucosa production exceed the cleaning capacity of the Eustachian tube (in cases of inflammation, for instance), then the mucosa waste may eventually accumulate in the middle ear (Sadé & Ar, 1997).
Moreover, the inside pressure of the middle ear needs to be precisely controlled. The isobaric system of the middle ear is thought to be composed of an afferent system transmitting the changes in the pressure gradient between the middle ear and the outside world, as well as an efferent system acting on key elements to restore the normal pressure when necessary (Salburgo et al., 2016). The pressure difference between the environment and the middle ear (which happens in planes at the take-off and landing, for instance) can be sensed by tympanic membrane mechanoreceptors (Pacini corpuscules) which are sensitive to the stretching of this membrane (Nagai & Tono, 1989) and lead to ear fullness. Other mechanoreceptors (Ruffini corpuscules) have been found outside the middle ear, near the Eustachian tube exit, namely, on the nasopharynx (Salburgo et al., 2016).
The gas composition in the middle ear is different from air and is very close to the gas composition of the venous blood in the middle ear mucosa. In particular, it presents a gas pressure “deficit” in oxygen, which is consumed by metabolic processes. This suggests that the middle ear is actually a poorly ventilated cavity and that the diffusive exchange with venous blood plays a greater role than the ventilation process permitted by the Eustachian tube opening. The middle ear pressure has therefore an inherent tendency to decrease due to gas exchange between the middle ear cavity and the venous blood. However, this decrease in pressure is compensated by the Eustachian tube ventilation allowing just enough gas to enter the middle ear to maintain the middle ear pressure at a physiological steady-state (Pau, Sievert, Just, & Sadé, 2009; Sadé & Ar, 1997). Any dysfunction or blockage of the Eustachian tube or changes of the middle ear mucosa would result in negative pressure in the middle ear and potentially to ear fullness.
Middle Ear Muscles
The stapedius muscle
The stapedius muscle (SM) is the smallest (∼1 mm) skeletal muscle in the human body. It is innervated by the facial nerve (seventh cranial nerve or CN VII). The SM has a protective role for the cochlea. Indeed, SM contraction, that is triggered by loud sounds, pulls on the stapes which result in a damping of the acoustic vibrations transmitted to the cochlea.
The tensor tympani muscle
The TTM is a 20-mm long skeletal muscle inserted between the cartilage of the Eustachian tube and the neck of the malleus (Aristeguieta, Acuña, & Ortiz, 2010). At the insertion point on the cartilage of the Eustachian tube, the TTM shares a tendon with the tensor veli palatini muscle (Kierner, Mayer, & Kirschhofer, 2002) suggesting these two muscles could be part of the same functional unit (see later). Muscle spindles that are part of the proprioceptive muscle organ have been found in man TTM (Kierner et al., 1999).
The TTM is innervated by the mandibular branch of the TGN (Mizuno et al., 1982). To note, the TGN also innervates the muscles of mastication (masseter, temporalis, lateral, and medial pterygoid), the tensor veli palatini, the mylohyoid muscle, and anterior belly of the digastric muscle. At the trigeminal motor nucleus, the TTM motor neurons receive diverse information from cortical regions and subcortical regions like cochlear nuclei (Billig, Yeager, Blikas, & Raz, 2007; Itoh et al., 1986; Rouiller, Capt, Dolivo, & De Ribaupierre, 1986).
Contraction of the TTM pulls the eardrum membrane inward and displaces the stapes into the vestibular canal (Hüttenbrink, 1989). The TTM contraction results in an increase in the impedance of the middle ear, thereby reducing sound transmission to the inner ear. In people with the ability of contracting voluntarily the TTM, it has been shown that this contraction can raise their hearing threshold of around 25 dB between 250 and 1 kHz (Angeli, Lise, Tabajara, & Maffacioli, 2013; Wickens, Floyd, & Bance, 2017). These results suggest that the main function of the TTM is to protect the ear against loud noises.
Interestingly, the TTM (and the SM) can be activated by many nonauditory stimuli such as sensory-motor activities in the region of the head and neck, for example, voluntary or involuntary eye closure; airflow to an eye socket; and tactile stimulation of the external auditory canal, speaking, yawning, swallowing, or chewing (Djupesland, 1965; Kim, Jang, Park, & Nam, 2015; Klockhoff, 1961; Lee et al., 2012; Ramirez et al., 2008; Salomon & Starr, 1963). The TTM may also be activated as part of the trigeminal nerve reflexes (corneal and blink reflexes, jaw opening and closing, and head retraction). This is consistent with its supposed protective role (Bradnam & Barry, 2013). The TTM (and the SM) contraction can also be triggered by the simple sight of a toy pistol (Djupesland, 1965). Interestingly, this anticipatory contraction was dependent on the level of subject anxiety (Westcott, 2006).
There is still another hypothesis on the functional role of the TTM. Based on the anatomical proximity of the tensor veli palatini muscle and TTM, it has been proposed that these two muscles form a functional unit controlling the middle ear pressure to maintain it equal to atmospheric pressure (Kierner et al., 2002). The tensor veli palatini muscle is well known for its role in opening the Eustachian tube (which communicates with nasal cavities), allowing for the tympanic cavity pressure to adjust to atmospheric pressure (see earlier). A difference in pressure between the auditory bulla and outside results in a distortion of the eardrum with in turn more or less strain on the TTM. The information concerning this strain could be sent to the brain via the TTM muscle spindle. The trigeminal motor root could consequently activate the tensor veli palatini muscle inducing the opening of the Eustachian tube allowing a readjustment of middle ear pressure (Duncan, 1982; Malkin, 1987). This hypothesis suggests that the TTM plays a sensory and signal transduction role in the middle ear. It also suggests that the pressure in the auditory bulla could be poorly regulated in patients with a dysfunctional TTM-tensor veli palatini muscle unit.
A Pathophysiological Model of TTTS
The pathophysiological model presented here is broadly influenced by the recent study of our group reporting the symptoms in a single but very well-documented case. According to this recent study, the symptoms triggered by an acoustic shock can be grouped in three different clusters (Londero et al., 2017). We use these symptom clusters as a framework for the present pathophysiological model of TTTS.
Acoustic Trauma: TTM Injury and Inflammation
It has been suggested that an acoustic shock (or trauma), potentially coupled to a particular emotional state, can cause a TTM hypercontraction (overuse) triggering a cascade of events leading to the symptom cluster mentioned earlier (Klockhoff, 1961; Klockhoff & Anderson, 1960; Westcott, 2006). The resulting effect of this hypercontraction could be even more important if it occurs in the case of TTM vulnerability (muscle fatigue, chronic hypoxia), namely, during times where the TTM is under particular strain due to overload, stress and noisy and loud environments. Call centers, where many cases of acoustic shock have been reported (Westcott, 2006), may combine all of these elements, including prolonged stress and strong focused auditory attention. The hypercontraction linked to an acoustic shock or trauma could lead to a more or less severe musculoskeletal disorder of the TTM, from a simple stiffening of the muscle to a more severe and pathologic condition such as tear, chronic, and spasmodic contraction. The feeling of ear fullness may result from the deformation of the tympanum detected by the mechanoreceptors inside the tympanic membrane (Densert, Ingelstedt, Ivarsson, & Pedersen, 1975; Londero et al., 2017; Mirza & Richardson, 2005; Nagai & Tono, 1989; Sakata et al., 2009; Yuasa et al., 1987) due to TTM contraction and the dysfunction of the TTM-tensor veli palatini muscle functional unit (Kierner et al., 2002).
The abnormal contraction of the TTM after a loud acoustic event may be similar and share some mechanisms with myofascial trigger point (MTrP). MTrP is defined as an hyperirritable spot in skeletal muscle associated with a contraction knot (“micro-cramp”) in a taut band (Arendt-Nielsen, Simons, & Pareja, 2007; Bron & Dommerholt, 2012; Fernández-de-las-Peñas, Cuadrado, Rocha & Sanchez, 2007; Shah et al., 2015; Simons, 2004; Teachey, Wijtmans, Cardarelli, & Levine, 2012). The MTrP has been described as having three components: a muscular component (contraction knot), a sensory component (pain), and an autonomic component (McPartland, 2004). One notes that spasmodic contraction of the TTM may also coexist with MTrPs. Although MTrPs concern only a small region of a muscle, spasmodic contraction is a violent contraction involving the whole muscle. The spasmodic contraction of the TTM may account for the feeling of fluttering ear that can be reported in some subjects after an acoustic shock and the spontaneous change in middle ear admittance (Klockhoff, 1976; Londero et al., 2017).
Injury of the TTM can be associated with many other adverse consequences. The main detrimental consequence of excessive and prolonged muscle contraction (muscle overload) is blood vessel compression. Importantly, this can result in a reduction in the local oxygen supply to the affected muscle. This phenomenon, in addition to a higher metabolic demand due to the prolonged contraction, can result in a reduction in the production of adenosine triphosphate (ATP) also called “ATP energy crisis” (Figure 1). In this circumstance, the muscle switches to an anaerobic glycolysis state to provide the muscle with adequate ATP. Lactic acid is then produced and accumulates in the muscle which increases the local acidity. This decrease in pH (increase of extracellular protons) can activate acid-sensing ion channels of nociceptors, thereby exciting these neurons (Frey Law et al., 2008; Gautam, Benson, & Sluka, 2010). A low pH can also downregulate acetylcholinesterase, increase the efficacy of acetylcholine, and maintain muscle contraction. Moreover, free Ca2+, needed for muscular contraction, has to return to the sarcoplasmic reticulum by the calcium pump for muscular relaxation. This process, however, is costly in ATP and cannot be properly done in case of severe energy depletion. The muscle thus stay contracted (until enough ATP is available), which could lead to further muscle injury (Bron & Dommerholt, 2012; Gissel & Clausen, 2001).
Schematic representation showing the cascade of events following an acoustic shock and leading to a deleterious and hard to treat vicious circle. The acoustic shock is thought to produce a hypercontraction of the TTM (muscle overuse), which may lead to TTM overload, hypoxia, and ATP-energy crisis. The muscle then switches to anaerobic glycolysis, which produces lactic acid and reduces the pH. The pH reduction can activate the nociceptors through the acid-sensitive ion channels, which in turn can result in an antidromic release of SP and CGRP. The mastocytes, activated by proinflammatory molecules, release histamine, thereby activating nociceptors in a slightly adjacent region, which can in turn trigger the release of SP and CGRP. This loop (interaction between histamine and proinflammatory molecules) is characteristic of the neurogenic inflammation. Finally, pain associated with the cascade of event may trigger a muscular protective reflex that may increase further the TTM overload. Finally, the activation of the TGN and TCC could generate the symptom cluster after an acoustic shock. The acoustic shock may also lead to stress or anxiety (fear of experiencing another acoustic shock) and attention focused on the auditory modality could modulate TTM motoneurons excitability (reduction of TTM contraction threshold) via the TCC. The state of stress or anxiety can also activate the sympathetic nervous system, which may amplify and maintain pain. Finally, musculoskeletal disorders in the head–neck region can be transmitted to the middle ear and the TTM via the TCC (that has bidirectional sensory-motor connections for the inputs coming from the head–neck region). TCC = trigeminal cervical complex; TGN = trigeminal nerve; CGRP = calcitonin-gene-related peptide; TTM = tensor tympani muscle; DCN = dorsal cochlear nucleus; SP = substance P; AChE = acetylcholinesterase.
The mastocytes can also migrate to the injured tissues and start to release histamine and platelet-activating factor leading to serotonin release from platelets. Once activated, the TTM nociceptors can release neuropeptides, namely, substance P (SP) and calcitonin-gene-related peptide (CGRP) antidromically at the injury (peripheral) site (Yamazaki & Sato, 2014). Interestingly, a study using an in vivo microanalytical technique showed that the concentration of protons, bradykinin, CGRP, SP, tumor necrosis factor-α, interleukin-1β, serotonin, and norepinephrine was higher in MTrPs compared with normal muscular region (Shah, Phillips, Danoff, & Gerber, 2005). The release of these substances can lead to a cascade of events, including degranulation of mastocytes, localized edema through increased permeability and dilation of blood vessels (Richardson & Vasko, 2002). The proinflammatory neuropeptides can equally diffuse to adjacent unaffected regions and induce the release of histamine by local mastocytes, which in turn also results in the release of SP and CGRP by nociceptive fibers. Interactions are thereby established between histamine and the algogenic neuropeptides that are typical of a neurogenic inflammation spreading to a distal nonaffected region (Rosa & Fantozzi, 2013). The neurogenic inflammation can also diffuse to the tympanum which is rich in mastocytes resulting in dilated radiating vessels in the pars tensa (Nagaraj & Linthicum, 1998).
Moreover, we speculate that the proinflammatory molecules released by the TTM lesion may diffuse to the middle ear mucosa and trigger strong inflammatory processes (Alm, Bloom, Hellström, Stenfors, & Widemar, 1983; Ebmeyer et al., 2005; Sadé & Ar, 1997). The middle ear mucosa has indeed all the ingredients that can trigger inflammation: mastocytes, nociceptors, and rich vascularization (Ebmeyer et al., 2005; Ylikoski & Panula, 1988). This inflammation might even lead to sterile otitis media (Juhn et al., 2008). The middle ear inflammation (such as any inflammatory processes) is associated with the secretion of many proinflammatory molecules and may lead to earache and otalgia (tingling and stabbing pain) as the middle ear mucosa is richly innervated with nociceptors (mandibular branch of the TGN) (Juhn, Jung, Lin, & Rhee, 1997). Middle ear inflammation can be associated with an increase of mucosa secretion and gas exchanges between the middle ear cavity and the richly vascularized mucosa epithelium (Ar et al., 2007; Juhn et al., 1997). In addition, the role of the Eustachian tube in preserving the middle ear pressure may be disturbed as the excess of mucus secreted under inflammatory conditions may block it. Finally, the gas exchange increase and the blockage of the Eustachian tube can both lead to negative pressure in the middle ear cavity. Sensed by the tympanic mechanoreceptors, this reduction of the middle ear pressure may contribute to the sensation of ear fullness (Ar et al., 2007; Sadé & Ar, 1997).
To note, the inflammatory molecules present in the middle ear cavity may cross the round window and cause inner ear damages (hearing loss), in particular in the high frequency region. One can further speculate that these molecules (such as ATP) (Bours, Dagnelie, Giuliani, Wesselius, & Di Virgilio, 2011; Cauwels, Rogge, Vandendriessche, Shiva, & Brouckaert, 2014) may further reach the organ of Corti and activate the unmyelinated type II afferent neurons synapsing with OHCs (Liu, Glowatzki, & Fuchs, 2015). These fibers are thought to be nociceptors signaling cochlear damages, by opposition of the type I fibers that transmit acoustic information. This hypothesis is inspired by the dichotomy in the somatosensory system between unmyelinated type C fibers signaling tissue damage and the myelinated fibers transmitting fine touch. The analogy between cochlear type II fibers and somatic type C fibers are reinforced by the fact that they are both activated by the proinflammatory molecules released during inflammatory processes and by ATP in particular (Basbaum, Bautista, Scherrer, & Julius, 2009; Cook & McCleskey, 2002; Liu et al., 2015). Finally, ATP and other proinflammatory molecules, produced in the middle ear during inflammatory processes and diffusing up to the Organ of Corti, may activate cochlear type II fibers and trigger sound-induced earache. One notes that ATP may also be released by the OHCs that are damaged due to the acoustic shock or trauma or due to the cytotoxic effects of proinflammatory molecules that diffused from the middle ear. Assuming that the diffusion of ATP secreted in the middle ear is limited to the cochlear base, sound-induced earache may be produced by high-frequency sound, which is consistent with informal patient reports.
Referred Pain
The trigeminal sensory inputs from regions of the head, including the middle ear, are collected by the trigemino-cervical complex (TCC) in the brainstem and then transmitted higher up in the central nervous system up to the cortex (Bradnam & Barry, 2013; Piovesan et al., 2003). The facial nerve, which is mainly motor, innervates the muscles of the face. Surprisingly, the facial muscles are devoid of proprioceptors and therefore lack proper proprioceptive feedbacks (Cobo, Solé-Magdalena, Menéndez, de Vicente, & Vega, 2017). However, several trigeminal nerve branches communicate with the main divisions of the facial nerve, thereby providing sensory innervation to the facial muscles. A TTM injury and the chronic inflammatory processes following an acoustic incident may be associated with neural hyperactivity of the trigeminal pathways and to the diverse feelings of pain felt in the ear by subjects following an acoustic shock or trauma (Londero et al., 2017; Westcott, 2006; Westcott et al., 2013; Figure 1).
We believe that the TCC plays a critical role in the pathophysiology of the symptom cluster triggered after acoustic shock or trauma (and other causes). The TCC receives convergent nociceptive and nonnociceptive afferents not only coming from the head region but also originating from the neck and the back of the head (Bartsch, 2005; Bartsch & Goadsby, 2003a; Figure 2). Importantly, sensory inputs to the spinal trigeminal nucleus and the upper spinal cord segments overlap. Indeed, wide dynamic range neurons of the spinal trigeminal nucleus in the TCC receive inputs from superficial and deep tissues and from nociceptive and nonnociceptive afferents of trigeminal and upper cervical spinal nerves (Mørch, Hu, Arendt-Nielsen, & Sessle, 2007; Figure 2). Moreover, the TCC has extensive connections to the somatosensory cortex, the primary motor cortex and brainstem motor regions, including the trigeminal motor neurons, facial motor neurons, and upper cervical motor neurons. This enables the TCC to indirectly modulate various spinal motoneurons excitability via multiple descending pathways (Bradnam & Barry, 2013). The trigeminal afferents and the TCC are also playing a role in articulating and controlling the sensorimotor reflex involving the head–neck complex, such as the eye blink reflex and the trigemino-cervical reflex (Di Lazzaro et al., 2006; Valls-Sole, 2012). In summary, the TCC is a key crossroad structure integrating sensory inputs coming from the head region, receiving modulatory influences from diverse areas, and projecting to diverse structures. This particular organization has two important consequences.
Schematic of the trigeminal pathway and the trigeminal cervical complex (TCC), with the mesencephalic nucleus (proprioception), the principal sensory nucleus (discriminative touch and proprioception), and the spinal sensory nucleus (pain and temperature). The face in the upper left of the figure shows the sensory areas innervated by the TGN and the C1-C4 spinal nerves (TGN: green: ophthalmic branch, orange: maxillary branch, blue: mandibular branch; C1-4 spinal nerves: light gray). The C1-C4 spinal nerves are all mixed nerves, that is, they transmit motor and sensory inputs between the spinal chord and the body. They innervate the back of the head and the neck region transmitting sensory (from skin, muscles—proprioception, tendons, and joints) and motor inputs. The C1-C4 spinal nerves also give rise to the suboccipital nerve (C1), the greater occipital nerve, also called Arnold’s nerve (C2), the third occipital nerve (C3), and the fourth occipital nerve (C4), which are all sensory. The spinal accessory nerves (cranial nerve XI) provide the motor command to the neck muscles (sternocleidomastoid and trapezius muscles). The spinal sensory nucleus contains wide dynamic range neurons that can be activated by inputs coming from different regions of the head and the neck (see zoom on the bottom right illustrating a wide dynamic range neuron receiving convergent inputs). The different nuclei of the TCC send also inputs to the trigeminal motor nucleus and the spinal accessory nucleus (providing inputs to the spinal accessory nerve, not shown). This particular connection pattern confers to the TCC bidirectional sensory-motor properties. Therefore, pain in the ear can produce referred pain in the neck and reversely. Furthermore, musculoskeletal disorders in the head–neck region may be transmitted to the muscles innervated by the trigeminal motor neurons (TTM or muscles of mastication), and reversely. TGG = trigeminal ganglion.
First, the functional convergence of TGN and C1-C4 inputs allows the bidirectional referral of painful sensations between the neck and the regions innervated by the TGN (Bartsch, 2005; Bartsch & Goadsby, 2002, 2003a, 2003b; Goadsby et al., 2017). An inflammatory soup applied on the dura matter has been shown to induce an increase in responsiveness of trigeminal second-order neurons to dural and cutaneous facial stimulation (Burstein, Yamamura, Malick, & Strassman, 1998). Moreover, convergent neurons at the level of C2 in the TCC showed an increased excitability to dural and greater occipital nerve stimulation after mustard oil-induced inflammation of the skin and muscles innervated by the greater occipital nerve (Bartsch & Goadsby, 2002). Another interesting study showed that experimentally induced pain in several masticatory and neck muscles can diffuse to remote regions of the head and neck. In particular, the temporalis posterior muscle (innervated by the TGN) and the sternocleidomastoid muscle (innervated by the C2-C3 nerves) can produce referred pain in a vast area of the head–neck complex (Schmidt-Hansen, Svensson, Jensen, Graven-Nielsen, & Bach, 2006). After an acoustic shock, subjects may report pain in the face and in the temporal and neck regions (Londero et al., 2017; Westcott, 2006). We suggest that these painful perceptions may be referred pain resulting from excitation of the wide dynamic range neurons in the spinal sensory nucleus of the TCC (Figure 2). On the other hand, it is also conceivable that injury or pain affecting neck region (cervical problems, neck injury after whiplash) or the manducatory apparatus (TMJDs) can produce referred pain in the ear (Bechter, Wieland, & Hamann, 2016; Jaber, Leonetti, Lawrason, & Feustel, 2008; Kuttila, Kuttila, Le Bell, Alanen, & Suonpää, 2004; Lam, Lawrence, & Tenenbaum, 2001; Michiels, Van de Heyning, Truijen, & De Hertogh, 2015; Ramirez et al., 2008; Riga, Xenellis, Peraki, Ferekidou, & Korres, 2010; Tranter & Graham, 2009; Wright & North, 2009).
Second, a normal or pathological contraction (musculoskeletal disorder, potentially associated with inflammation) of any muscle in the head–neck complex can activate, modulate activity, or decrease contraction threshold of other muscle in this region via the bidirectional properties of the TCC (Bradnam & Barry, 2013; Figure 2). Therefore, any muscle contraction in the region of the head or the neck region may influence the TTM, by reducing the contraction threshold or triggering contraction. In pathological circumstances, this mechanism may account for the otic symptoms that can be associated with musculoskeletal disorders affecting the head and neck region (Bechter et al., 2016; Bjorne, 1993; Ferendiuk, Zajdel, & Pihut, 2014; Lam et al., 2001; Levine, 1999; Ramirez et al., 2008; Riga et al., 2010; Rocha & Sanchez, 2007; Teachey et al., 2012). It is interesting to note that office workers using screen often report musculoskeletal disorders in the region of the neck and shoulders (Sauter, Schleifer, & Knutson, 1991). One notes that the development of musculoskeletal disorders in the region of the head–neck complex in subjects working in call centers may increase their susceptibility to acoustic shock.
It has been shown that voluntary contractions of head and neck muscles may be accompanied by TTM contraction. Importantly, the concomitant contraction of head or neck muscles and TTM may actually result from some form of pathology (Salomon & Starr, 1963). Consistent with the latter study, we reported that voluntarily (but not forcefully) eye blinking made by a subject exposed to an acoustic shock changed dramatically the middle ear admittance (Londero et al., 2017). The large effect of eye blinking on the middle ear admittance suggested that the admittance change resulted from the TTM contraction (the admittance change related to SM contraction alone is much smaller—Kim et al., 2015). This phenomenon has been described in the literature is called the forced eyelid closure syndrome and can be associated to tinnitus (Kaffenberger, Mandal, Schaitkin, & Hirsch, 2017; Kim et al., 2015; Lee et al., 2012; Ohki & Kato, 2012; Rock, 1995). The mechanisms of the forced eyelid closure syndrome are unclear. Interestingly, it has been reported that the forced eyelid closure syndrome can be triggered soon after wearing eye lenses (Lee et al., 2012). The latter result suggests that chronic activation of the TGN (in that case due to the eye lenses stimulating corneal receptors) may trigger some kind of neural plasticity (central sensitization, see later). This plasticity, possibly at the level of the spinal trigeminal nucleus where initially silent synapses may become unmasked, can then facilitate the reflex loop between the trigeminal sensory inputs and the trigeminal motor outputs (via the trigeminal motor nucleus; Figure 3).
Schematic on the putative connection between the sensory branch of the trigeminal nerve (ophthalmic division), the spinal trigeminal nucleus, the facial nucleus, and the trigeminal motor nucleus. In some abnormal circumstances potentially leading to central plasticity and unmasking of silent synapses (see text), the activation of the corneal receptors and the trigeminal nerve may activate TTM motoneurons, thereby triggering TTM contraction. The TTM contraction can be objectified measuring middle ear admittance (Londero et al., 2017).
In addition to the bidirectional facilitation between the different sensorimotor areas of the head–neck complex exerted through the TCC, descending projections from central regions including the cortex can also modulate the TCC. This top-down modulation may influence the muscle contraction threshold during stress or anxiety (Bradnam & Barry, 2013). Namely, stress or anxiety may be associated with a reduced TTM contraction threshold. One notes that these top-down modulatory influences on TTM via the TCC may account for the controversy around the ability of loud sound to trigger TTM contraction (Djupesland, 1965; Jones, Mason, Sunkaraneni, & Baguley, 2008; Klockhoff, 1961; Stach, Jerger, & Jenkins, 1984). In other words, TTM contraction may be triggered by loud sounds only under particular emotional context. In the same vein, it has been shown that contralateral acoustic stimulation can produce TTM contraction if the sound is able to elicit a cochleo-palpebral reflex (Djupesland, 1965).
Otalgia and Autonomic Symptoms
The autonomic nervous system may also be involved in the cascade of events leading to muscle overload and pain. The activation of nociceptors present on the TTM may evoke a motor and sympathetic reflex, in the same way as nociceptive signals can trigger protective motor reflexes, such as flexor reflex withdrawal, for instance. These types of reflexes may, however, have the drawbacks of increasing abnormal muscle contraction and ultimately generate pain. Indeed, reflex muscle contraction, which can activate muscle nociceptors, may in turn enhance the motor reflex, thereby creating a “vicious circle” (Grassi & Passatore, 1988). Antagonists of α1-adrenergic receptors have been shown to decrease the spontaneous electric activity that is elevated in MFTrPs (Chen, Chen, Kuan, Chung, & Hong, 1998; Rivner, 2001). Interestingly, intravenous administration of an autonomic nerve blocker, known as having an antispastic effect, can reduce the sensation of aural fullness, improve hearing in low frequencies, and reduce tinnitus and stiffness in the neck and shoulder (Yuasa et al., 1987). It is possible that the autonomic nerve blocker produced the relaxation of the TTM, thereby improving symptoms associated with TTM tonic contraction or spasms.
The chronic pain, associated with tissue inflammation, may be maintained and even amplified by the sympathetic nervous system (Baron, Levine, & Fields, 1999; Baron, Schattschneider, Binder, Siebrecht, & Wasner, 2002). This is supported by studies showing that under certain pathological conditions (inflammation), noradrenaline (or norepinephrine) can activate primary nociceptors (Hu & Zhu, 1989; Roberts & Elardo, 1985), thereby triggering or increasing pain (Baron et al., 2002). Interestingly, others have reported improvement in the pain condition after blocking the sympathetic nervous system (Price, Long, Wilsey, & Rafii, 1998). More so, prostaglandins, released by the postganglionic neurons, may sensitize the nociceptors (Gonzales, Goldyne, Taiwo, & Levine, 1989). It has been suggested that the interaction between the postganglionic neurons of the sympathetic nervous system and the primary afferent nociceptors is mediated by α2-adrenoceptors (Sato, Suzuki, Iseki, & Kumazawa, 1993).
Finally, it has been reported that MFTrPs located in the region of the head and neck may cause lacrimation, salivation, and nasal discharge, fluids secreted by the glands that are under the control of the parasympathetic system (McPartland, 2004). The TCC has a reflex connection with the parasympathetic system, which arises from the superior salivatory nucleus and distributes fibers to the head region via the sphenopalatine ganglion. This reflex loop between the TGN and the parasympathetic pathway has been suggested to account for the autonomic symptoms associated with trigeminal-autonomic cephalalgia (Bartsch & Goadsby, 2003a; Eller & Goadsby, 2016). We propose that the activation of the trigeminal-autonomic reflex may account for the nasal congestion, rhinorrhea, and the tympanic hyperemia sometimes reported after an acoustic shock (Londero et al., 2017; Figure 4).
Schematic accounting for the symptom cluster triggered by an acoustic shock. The acoustic shock may lead to TTM overuse and injury and inflammatory processes. This activates the trigeminal pathway which is associated with pain and tinnitus (potentially through modulating activity of DCN neurons or stria vascularis). The activation of the TGN can activate the parasympathetic pathway through the parasympathetic-trigeminal reflex. This may explain the autonomic symptoms such as blocked nose and tympanum hyperemia. Moreover, the wide dynamic range neurons of the TCC integrate inputs from the head–neck complex, which may account for referred pain in the temporal and neck region. Finally, the sympathetic system may maintain and amplify pain by interfering with the nociceptor activity. ACC = anterior cingulate cortex, PH = posterior hypothalamus, LC = locus cœruleus, PAG = periaqueductal gray, CN = cochlear nucleus, TGG = trigeminal ganglion; TGN = trigeminal nerve.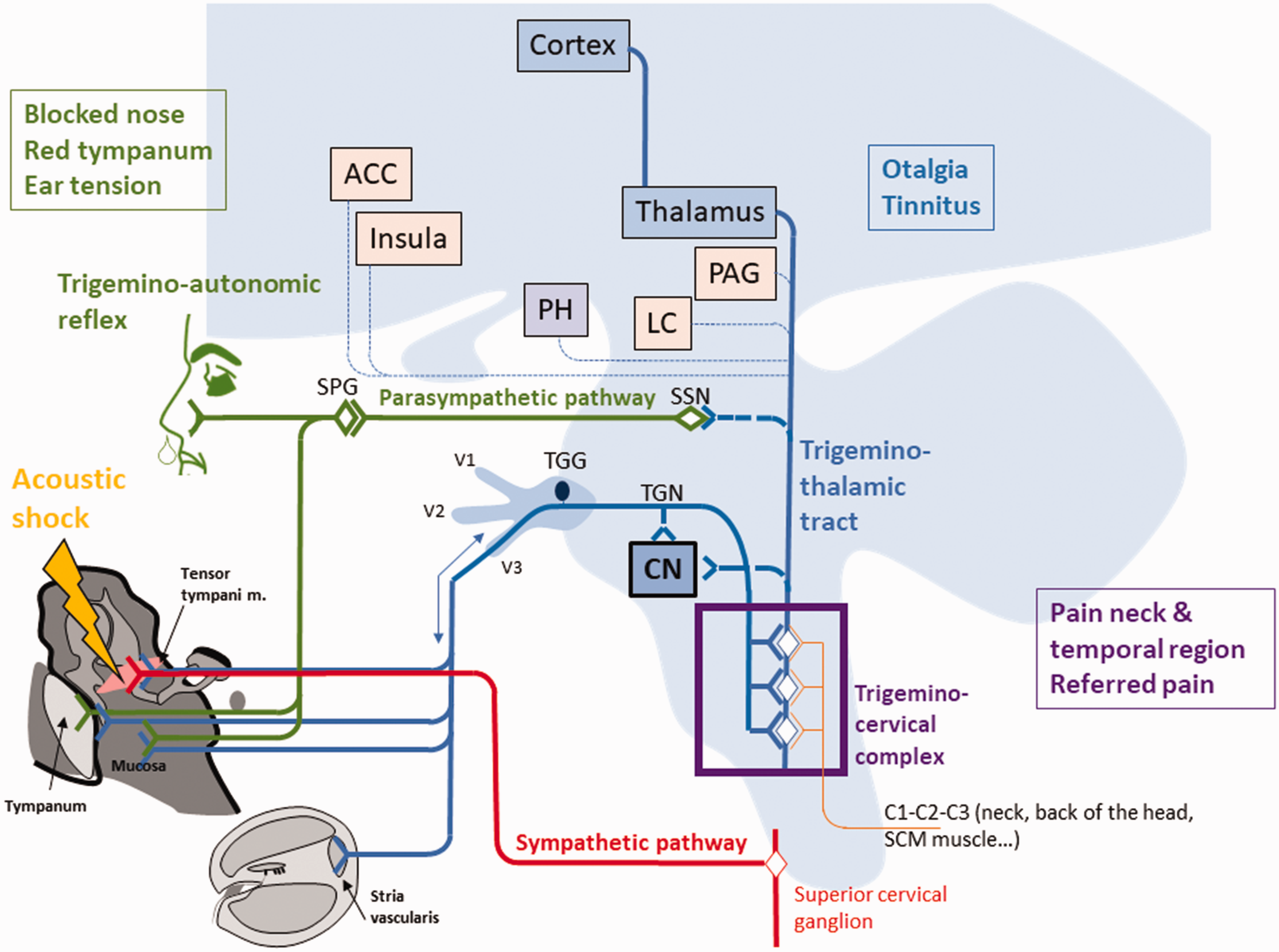
Tinnitus
It still remains unclear how tinnitus can result from acoustic shock or trauma, that is, the tinnitus subtype associated with the symptom cluster described earlier. Tinnitus can appear immediately or a few days or weeks after the acoustic trauma (Londero et al., 2017). The delay between the acoustic trauma and the emergence of tinnitus suggests that the tinnitus-related mechanisms can build up over time, which is consistent with the vicious circle shown in Figure 1.
Cochlear damages
It has been shown that brief and moderate-to-intense sound can damage OHC stereocilia (Liberman & Dodds, 1984, 1987; Wang, Hirose, & Liberman, 2002). This specific lesion may affect the mechano-electric transducer (MET) channels by reducing their opening probability. At equilibrium, 50% of these channels are open allowing a relatively important current shunt through them. The MET channels opening probability therefore has an impact on endocochlear potential through the size of this current shunt. When the probability of opening is reduced, the current shunt is reduced and the endocochlear potential is increased (the opposite is true when the opening probability is increased). The increase in endocochlear potential could have two consequences: (i) an increase in cochlear nerve spontaneous activity (through the depolarization of inner hair cells; Sewell, 1984) and (ii) a K+ ions increase in the cochlear duct leading to the influx of water into the cochlear duct through aquaporins thus causing an endolymphatic hydrops (Salt, 2004). Tinnitus that emerges after brief and intense acoustic trauma may result from this mechanism. This mechanism (reduction in MET channel opening probability) may also be at work during spasmodic contraction of the TTM (producing low-frequency vibrations) or when the auditory system is stimulated by very low frequencies (Drexl et al., 2014; Noreña, 2015; Salt & DeMott, 1999).
TGN modulation
Tinnitus that follows an acoustic trauma has been reported to fluctuate over time, in close correlation with tingling in the ear and otalgia (Londero et al., 2017). We have suggested that pain in the ear results from TGN activation due to TTM overload and injury (see earlier). Interestingly, the TGN innervates the cochlea and the vestibular labyrinth: the blood vessels around the spiral modiolus, the stria vascularis, and the dark cell region of the cristae ampullaris (Vass et al., 1997, 2001; Vass, Shore, Nuttall, & Miller, 1998). The action of TGN on the inner ear may contribute to generating tinnitus or modulate tinnitus loudness. The antidromic activation of the TGN ganglion may change vascular permeability in the inner ear. This may affect the composition of the endolymph (i.e., increase of the K+ concentration) secreted by the stria vascularis and the dark cells, which may then lead to tinnitus and other disorders (hearing disorders and vertigo; Vass et al., 2001). Furthermore, this mechanism may also account for hearing and vestibular disorders that can accompany migraine (Langguth, Hund, Landgrebe, & Schecklmann, 2017; Vass et al., 2001).
Furthermore, the TGN projects broadly to the central auditory system (Dehmel, Cui, & Shore, 2008; Shore, 2005; Shore, Vass, Wys, & Altschuler, 2000; Zhou & Shore, 2004, 2006). Electrical stimulation of the TGN can change the firing activity of neurons in the dorsal cochlear nucleus (DCN; Shore, Koehler, Oldakowski, Hughes, & Syed, 2008). Interestingly, noise-induced hearing loss increases the excitatory effects of TGN activation on neural activity. Moreover, the neurons that are activated by TGN stimulation are also showing enhanced spontaneous activity after noise-induced hearing loss (Shore et al., 2008). These results suggest that the excitatory influence of the TGN on neural activity in the dorsal cochlear nucleus can be involved in the generation or the modulation of tinnitus (Wu, Stefanescu, Martel, & Shore, 2015).
In summary, the TGN activated or modulated by TTM contraction or injury and the middle ear inflammation may play a critical role in the generation or modulation of tinnitus (Bechter et al., 2016; Rocha & Sanchez, 2007; Teachey et al., 2012; Tranter & Graham, 2009). Tinnitus may result from the TGN impact directly on the cochlea (stria vascularis) or on DCN neurons in the central auditory system (Shore et al., 2008; Vass et al., 1998). The rapid modulation of tinnitus loudness during somatic maneuvers or cervical manipulations suggests that “somatic” tinnitus is related to the modulation of neural excitability at central level (on the DCN) rather than at the cochlear level (Levine, 1999; Levine, Abel, & Cheng, 2003; Teachey et al., 2012).
Peripheral and Central Sensitization
We have described earlier that TTM overload potentially induced after noise trauma may lead to the local release of proinflammatory molecules and “sensitizers,” such as prostaglandins or SP, causing peripheral sensitization of nociceptors. This peripheral sensitization can increase the spontaneous firing of nociceptors and lower the action potential firing threshold, which may result in hyperexcitability and hyperalgesia (Fernández-de-las-Peñas et al., 2007).
Moreover, the continuous barrage of nociceptive signals coming from the TTM and the middle ear, enhanced by peripheral sensitization, may eventually cause central sensitization in the TCC. The central sensitization can further increase neural responsiveness or what is sometimes called the “central gain” (Woolf & Salter, 2000). In this case, the low threshold nonnociceptive fibers can become activated by stimulation that would be normally subthreshold. This phenomenon can account for allodynia (nonnociceptive stimulation producing pain). Moreover, above and subthreshold nociceptive inputs are equally amplified leading to hyperalgesia (amplification of nociceptive information) and a spreading of the nociceptive hypersensitization to regions beyond the injury (Ali, Meyer, & Campbell, 1996; Kilo, Schmelz, Koltzenburg, , & Handwerker, 1994). The central sensitization occurring at the level of the TCC may also contribute to and even enhance the bidirectional referral of painful sensations, the concomitant contraction of head and neck muscles and the TTM (see earlier), and the bidirectional “transmission” of musculoskeletal disorders between the neck and the head (Bartsch & Goadsby, 2002; Burstein et al., 1998; Gola, Chossegros, Orthlieb, Lepetre, & Ulmer, 1992; Lee et al., 2012; Londero et al., 2017; Salomon & Starr, 1963; Tranter & Graham, 2009).
Finally, peripheral and central sensitization of the TGN pathways may contribute to shifting the pathophysiology triggered after an acoustic shock from acute to chronic, by maintaining and amplifying a vicious circle between TTM injury and the TCC activation and feedback. Once these sensitization processes are involved, pain can persist in the absence of the injury and can be very long and hard to treat. One notes that the mechanisms of central sensitization may also play a role in the generation of tinnitus, in particular in the DCN where nonauditory inputs can be enhanced after cochlear damages (Shore et al., 2008; Wu et al., 2015).
Exiting the Loop
We have described earlier that all symptoms following acoustic shock or trauma may result from a cascade of events initially triggered by TTM overuse, overload, and ultimately injury. Pain linked to inflammation due to tissue damage generally stops once the tissue has repaired itself. Skeletal muscles have the remarkable ability to regenerate themselves after an injury. This ability depends on the presence, differentiation properties, and proliferation of myoblasts, the muscle precursors (Pavlath et al., 1998). The repair of TTM after the lesion may therefore restore a normal middle ear function and abolish all symptoms. However, masseter muscles have a limited ability to repair after damage (Dessem & Lovering, 2011; Pavlath et al., 1998). Considering the common embryologic origins of masseter muscles and the TTM, the latter might also have a limited ability for repair. This characteristic might explain its particular vulnerability to repeated harm from the acoustic environment and why relapse is generally much more severe than the initial acoustic shock (Milhinch, 2001). As suggested earlier, the TTM may be part of the trigeminal reflexes. Importantly, a prolonged emotional stress or anxiety due to the acoustic shock (or other causes) may contribute to lowering the threshold of sound-induced TTM contraction and maintain this threshold at a low level (Djupesland, 1965; Klockhoff & Anderson, 1960; Westcott, 2006; Figure 2). It is noteworthy to note that the TTM has a serotonergic innervation that could contribute to a TTM modulation by an emotional state (Thompson, Thompson, & Britton, 1998). The threshold reduction in the sound-induced TTM contraction due to psychological factors (including auditory attention) may then contribute to aggravate the symptoms and slow down the recovery process by preventing the TTM to rest.
In place or in addition to the spontaneous recovery of TTM, several clinical approaches may be used to improve a patient condition. One strategy involves incapacitating the TTM by severing it. This treatment has been used experimentally in Ménière’s disease. This surgery results in an improvement in perception thresholds, tinnitus, feeling of ear fullness, and vertigo (De Valck, Van Rompaey, Wuyts, & Van de Heyning, 2009; Franz, Hamzavi, Schneider, & Ehrenberger, 2003; Loader, Beicht, Hamzavi, & Franz, 2012, 2013). Pharmacological approaches, such as the use of muscle relaxants or anticholinergic agents (botox), could also be considered to reduce TTM contractions (Yuasa et al., 1987). Methods using analgesics or anti-inflammatory agents could be efficient. Moreover, muscle tone and pain being possibly under the modulation of the sympathetic nervous system, blocking the sympathetic nervous system may improve the symptom cluster (Baron et al., 1999; Fernández-de-las-Peñas et al., 2007; McPartland & Simons, 2006). An autonomic nerve blocker administered intravenously has been shown to reduce the sensation of aural fullness, which is possibly related to TTM contraction (Yuasa et al., 1987). Interestingly, in a single case study, Westcott et al. (in preparation) reported that stellate ganglion blockage improved and even completely suppressed the sound-induced pain seen after an acoustic shock.
In subjects with muscular or other problems, such as MTrP or TMJD, it would appear as common sense to treat first and foremost the cause of the problem (Ferendiuk et al., 2014). On a similar note, if subjects suffer a combination of functional disorders, a global treatment for all problems is required to increase the treatment efficiency. In this respect, it has been shown that joint treatment of TMJD and cervical issues reduces tinnitus, vestibular dysfunction, feeling of ear fullness, and pain (Bjorne, 1993; Björne, 2007; Bjorne & Agerberg, 2003). Globally, all methods (e.g., pharmacological or other —such as muscle stretching) leading to the decreased diffusion of spasmodic activity to the TTM by (the) affected muscle(s) would be beneficial (Björne, 2007).
Vernon suggested providing subjects with hyperacusis an electronic device aimed at limiting loud sounds while amplifying soft sounds (Vernon, Fenwick, & Nunley, 2002). This device limits the possibility to experience noise hazards while not overprotecting the ear. This type of device can maintain a certain level of stimulation that may be required for desensitizing the auditory system or prevent an increase of central gain (Formby, Sherlock, & Gold, 2003; Noreña, 2011, 2015; Noreña & Chery-Croze, 2007; Noreña & Farley, 2012). At the same time, it may reduce anxiety related to noise hazards, thereby allowing for a more relaxed life. Indeed, the emotional nervous system likely modulates the level of the TTM contraction (Djupesland, 1965; Thompson et al., 1998; Westcott, 2006). It is therefore probable that behavioral approaches for stress and anxiety reduction, such as relaxation or behavioral and cognitive therapies (Cima, Andersson, Schmidt, & Henry, 2014; Guastella, Mick, & Laurent, 2008) can affect TTM tonicity and triggering threshold. By promoting TTM relaxation, these methods could contribute to minimize or even abolish TTTS symptoms.
Finally, due to the progressive centralization of the pathophysiology after an acoustic shock, it is possible that severing or resting the TTM does not always reduce chronic pain after acoustic shock (Westcott, 2016). In all cases, it is preferable to act quickly on the local damage in order to stop inflammatory cascades associated with a TTM injury to prevent peripheral and central sensitization leading to functional disability.
Conclusion
We developed a model accounting for the symptom cluster associated with an acoustic shock or trauma. The triggering event is the overuse of the TTM, which could be associated with a TTM injury of varying severity, possibly leading to TTM chronic contraction or spasms. The initial TTM wound causes pain, which can spread through inflammatory processes, up to the middle ear mucosa, and may be amplified and persist beyond the tissue damage when peripheral and central sensitization mechanisms are at stake. The main TGN relay in the brainstem is the TCC, a crossroad structure that integrates sensory information from the head–neck complex. The broad integration of the TCC may account for referred pain outside the ear when the middle ear is injured. The sympathetic nervous system may also be involved in maintaining and amplifying pain, while the trigeminal-parasympathetic reflex may account for autonomic symptoms such as blocked nose and tympanum hyperemia. Finally, tinnitus may be modulated and result from the excitatory modulation exerted on the central auditory system or on the cochlea (stria vascularis) by the TGN activation. The present model is testable: In particular, TTM overload may be revealed by middle ear impedancemetry and middle ear inflammation by otoscopy or tympanocentesis, for instance.
Footnotes
Acknowledgments
The authors would like to thank Stéphane Gallego, Brahim Tighilet, Luis Garcia-Larrea, Anne Donnet, and Marina Siponen for their useful comments on earlier versions of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been funded through the financial assistance of CNRS, Aix-Marseille Université, B2V, and Klesia.