
Abstract
Sodium alginate fibers have been extensively studied due to being non-toxic and have high moisture retention, high oxygen permeability, biocompatibility, and biodegradability. However, their application has been limited due to their poor mechanical performance. In this study, poly(ethylene glycol) diglycidyl ether-modified sodium alginate fibers were prepared by spinning the solution after mixed reactions through a spinneret into a coagulation bath containing aqueous CaCl2. The properties of the spinning solution, the structure and physical properties of the modified sodium alginate fibers with various poly(ethylene glycol) diglycidyl ether contents were investigated. A poly(ethylene glycol) diglycidyl ether content of the modified sodium alginate fibers of 15 wt% gave optimal breaking strength and elongation at break, improving them by 78 % and 114 %, respectively, compared with pure sodium alginate fibers. The thermal stability of the modified sodium alginate fibers was also improved.
Keywords
Introduction
Sodium alginate (SA) has drawn more and more attention in recent years due to its biodegradability, nontoxicity, environmental benefits, biocompatibility, and low cost.1,2 It is a renewable and sustainable biopolymer that is extracted from brown seaweeds.3–5 Since it was discovered in 1881, alginate has been widely used in food, wound management, tissue engineering, water treatment, textiles, and many other industries.6–16 SA is a natural polysaccharide, which consists of β-(1–4)-linked d-mannuronic acid (M) residues and α-(1–4)-linked l-guluronic acid (G) residues. 17 These two residues are distributed randomly and form M, G, and MG blocks within polymer chains depending on different compositions of M and G residues.18–20 Through ionic bonding with multivalent cations (such as Ca2+), the G segments of SA can be gelated and the phase of SA transformed to water-insoluble calcium alginate. 21 Moreover, there are a number of carboxylate and hydroxyl groups in SA polymer chains, so it is easy to prepare SA-based functional derivatives.22–24 Recently, researchers have been paying more attention to the bio-based textiles and fibers due to textile waste pollution. SA-based fibrous materials have many potential applications.25,26
SA fibers are bio-based fibers usually formed from the wet spinning of an aqueous solution of SA in a calcium chloride coagulation bath. During the spinning process, a special egg-box structure is formed between the G block and Ca2+, strengthening the connection among alginate polymer chains, turning alginate into a gel, and dehydrating the gel to form fibers.27–29 However, low mechanical strength is a major problem for the further use of SA fibers in various fields. To improve the mechanical strength of SA fibers, physical blending and chemical modification are two effective methods.30,31 A second component is usually added into the SA matrix to improve the performance of SA fibers. A series of materials, such as carbon nanotubes, graphene, cellulose nanocrystals, and polyvinyl alcohol have been used to improve the mechanical properties of SA fibers.32–35 Compared with physical blending, chemical modification is usually achieved by modifying the polymer chain structures of SA with crosslinking agents because there are many carboxylic groups and hydroxyl groups in SA chains. 36 Poly(ethylene glycol) diglycidyl ether (PEGDE) is widely used in biomedical and biomaterial fields as a novel environmentally friendly crosslinker. Under certain conditions, the two epoxy end groups of PEGDE are vulnerable to the nucleophilic attack of anionic polysaccharide and can form ether-crosslinked bonds with the hydroxyl and carboxyl groups of polysaccharides through etherification. Hence, PEGDE is widely used for the crosslinking of oligosaccharides and polysaccharides. Under alkaline conditions, Kono et al. 37 successfully prepared carboxymethyl cellulose (CMC)-based novel hydrogels and insoluble cyclodextrin polymers (CDPs) using PEGDE as a crosslinker. Zhu et al. 38 prepared silk fibroin (SF)/SA composite fibers using PEGDE as a crosslinking agent to enhance the breaking strength of the composite fibers.
In this study, modified SA fibers were prepared through the chemical crosslinking of PEGDE and SA with various PEGDE contents. The flowing property of the spinning solution, Fourier transform infrared (FTIR) spectrograms, microstructures, and mechanical and thermal properties of the modified SA fibers were examined.
Materials and Methods
Chemicals
SA (industrial grade) was purchased from Hai Zhi Lin Biotech LLC, Qingdao, China. PEGDE (99%) was procured from Adamas-Beta Corp., Shanghai, China. Anhydrous calcium chloride (CaCl2, analytical grade) was supplied by BASF Corp., Tianjin, China.
Preparation of Modified SA Fibers
Spinning solutions with various PEGDE-to-SA mass ratios (0, 10, 15, 20, 25, and 30 wt%) were prepared. The solution volume and SA concentration were 100 mL and 4 wt%, respectively. The spinning solution was mechanically stirred for 5 h at 600 r/min at 60 °C to completely mix PEGDE with SA and then allowed to settle to remove bubbles after the reaction. The solution was extruded from a self-constructed wet spinning machine at 15 m/min. The spinning solution was solidified in a coagulation bath with 5 wt% CaCl2. The modified SA fibers were finally obtained after subsequent stretching, washing, and alcohol dehydration.
Characterization
An MCR 301 Rheometer (Physica Pear Corp., Austria) was used to test the apparent viscosity and rheological properties of the spinning solutions according to ASTM D3795-00a(2012). The shear scanning mode with a temperature of 30 °C, a scanning rate of 1 rad/s, and a strain of 5 % was used. The viscosity of the spinning solution was recorded every 30 s. The spinning solution with various PEGDE contents was dropped on a DV2T viscosity meter (Brookfield Corp., USA) to measure its viscosity at a temperature of 25 °C, a spinning rate of 3 r/min, and a shear rate of 0.84 s−1.
A Nicolet 5700 FTIR spectrometer (Thermo Nicolet Corp., USA) was used to characterize the chemical structure of the modified SA fibers according to GB/T 32199-2015. The scanning range, resolution, and scanning cycle values for the analysis were set to 500–4000 cm−1, 2 cm−1, and 32, respectively. An AVANCE III HD nuclear magnetic resonance (NMR) spectrometer (Bruker Corp., Germany) was also used to characterize the chemical structure of modified SA fibers. The spinning frequency, delay time, contact time, spectral width, collecting time, and field strength for the analysis were 8 kHz, 2 s, 2 ms, 62.5 kHz, 4.74 ms, and 9.4 T, respectively.
A JSM-6700F scanning electron microscope (SEM; JEOL, Japan) was used to observe the micromorphologies of the cross section and surface of modified SA fibers according to GB/T 17359-2012. The fibers were treated by gold spraying and the accelerating voltage was 15 kV.
The middle-cut method was adopted to measure the linear density (Hastelloy Slicer; Zhongxian Instrument LLC, China) of modified SA fibers at a temperature of 20 °C and a relative humidity (RH) of 65%. Ten measurements were conducted for each sample and the mean value was reported. According to the regulations specified by GB/T 14337-2008 Testing Methods for Tensile Properties of Man-made Staple Fibers, an LLY-06E electronic single fiber strength tester (Laizhou Electronic Device, China) was used to measure the fiber mechanical performance.
A TG209 F3 thermalgravimetric analyzer (NETZSCH, Germany) was used to measure the thermal stability of modified SA fibers under the protection of nitrogen according to ASTM E2402-11(2017). The temperature elevating rate was 10 °C/min and the temperature range was 30 °C–800 °C.
Results and Discussion
The viscosity of the spinning solution as a function of time during the reaction between SA and PEGDE is shown in Figure 1a. It is noticeable that the viscosity of the SA solution did not vary greatly with the increasing time. However, when PEGDE was added in the SA solution, the viscosity of the spinning solution increased with the prolonged reaction time. Moreover, as the PEGDE contents increased, the viscosity of the solution first increased and then decreased.
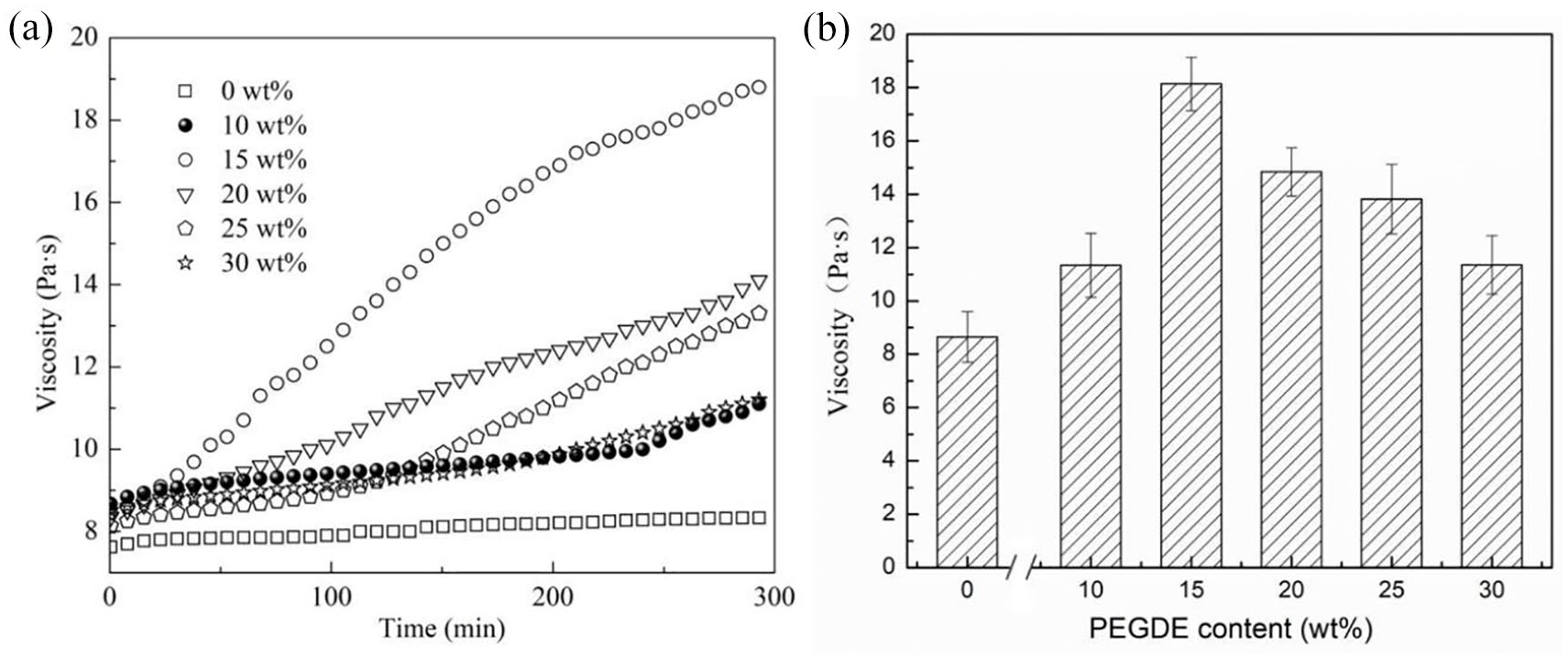
(a) Flowing property of the spinning solution with various PEGDE contents at different times and (b) the viscosity of the modified SA spinning solution with various PEGDE contents.
The addition of PEGDE increased the viscosity of the SA solution, demonstrating that PEGDE and SA reacted under neutral conditions. Figure 1b shows the viscosity of the spinning solution after the reaction of SA and PEGDE. The obtained data corresponded well to those in Figure 1a. It is noticeable that the viscosity of the spinning solutions with different PEGDE-to-SA mass ratios first increased and then decreased. When the PEGDE-to-SA mass ratio was 15 wt%, the viscosity of the solution reached the maximum value, indicating that etherification occurred between SA and PEGDE. When the PEGDE content was low, fewer crosslinking points existed during the reaction; thus, the viscosity of the spinning solution was only influenced slightly. As the PEGDE contents increased, the density of crosslinking points increased. Hence, a denser crosslinked network structure was formed, which restricted the relative movement of polymer chains and increased the viscosity. Flexible molecular chains of PEGDE acted as bridges among alginate polymers that were not crosslinked with Ca2+. When the PEGDE contents reached a certain level, PEGDE increased the distance among polymer chains, reduced the possibility of entanglement point formation among molecular chains, and reduced the viscosity. 39
The ring-opening reaction of PEGDE could occur easily since it contains a large number of ether bonds. Alginate anions in the SA aqueous solution attacked the carbon atom in the PEGDE end epoxy groups. Due to the high tension, energy, and reactivity of the epoxy structure, nucleophilic substitution occurred between PEGDE and the carboxyl groups of SA, forming a crosslinked network structure. Figure 2a displays the FTIR spectra of the modified SA fibers. A wide peak at 3300 cm−1 corresponding to the stretching vibration of the carboxyl groups in SA molecules was observed in the spectra of all fibers. The peaks at 2924, 1600, and 1416 cm−1 appeared from the stretching vibration of saturated –CH, and the asymmetrical and symmetrical stretching vibration of –COO, respectively. The stretching vibration adsorption peak of the β-1,4-mannuronic (C-O-C) bond connecting two blocks was observed at 1024 cm−1. The peaks at 841 and 1251 cm−1 are characteristic of the epoxy groups and the peak at 2866 cm−1 is the characteristic adsorption peak of –CH2. No PEGDE epoxy characteristic adsorption peaks existed in the spectra of modified SA fibers, indicating that the epoxy rings of PEGDE had all reacted. However, two new adsorption peaks at 2921 and 2851 cm−1 appeared in modified SA fibers (Figure 2b). These new peaks were associated with the asymmetrical and symmetrical stretching vibration of –CH2, demonstrating the reaction between PEGDE and SA.
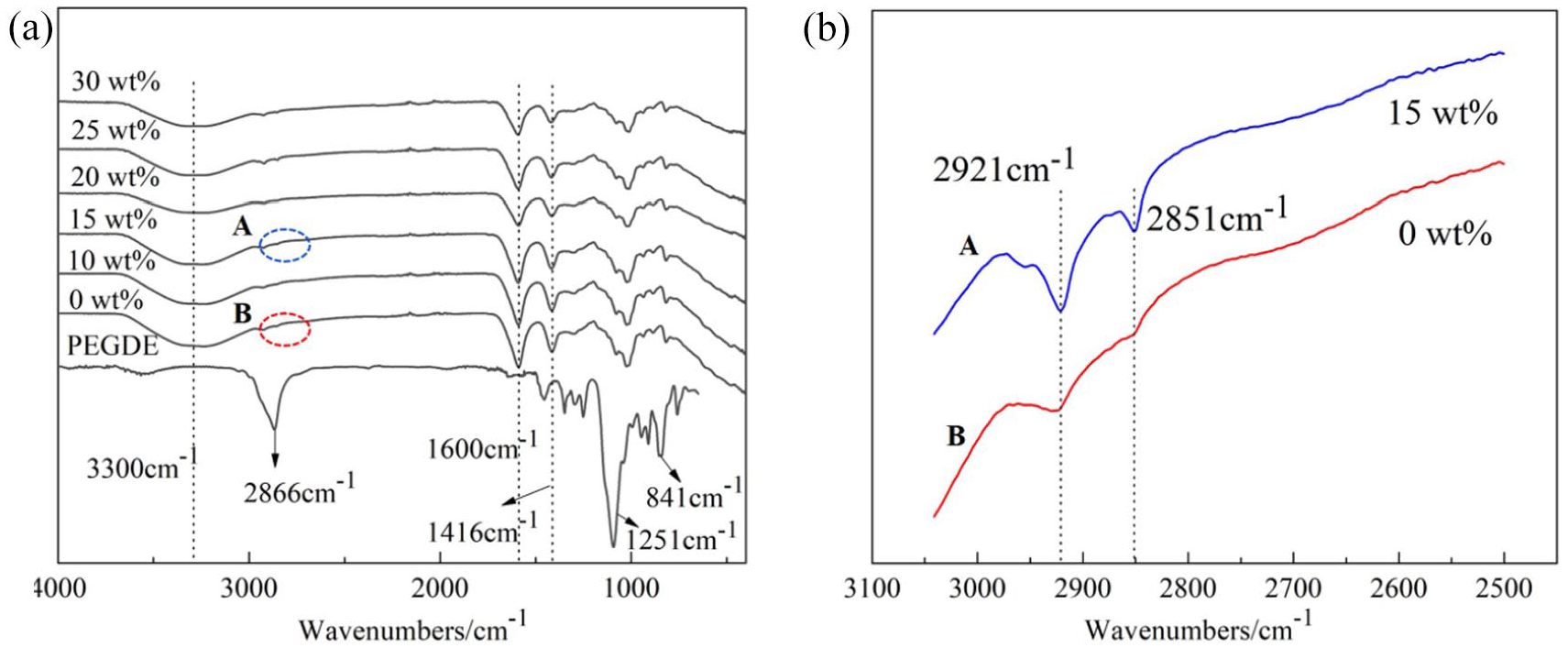
(a) FTIR spectrograms of modified SA fibers with various PEGDE contents and (b) the magnification peaks of FTIR spectrograms A&B in (a).
The 13C NMR spectra of liquid PEGDE are presented in Figure 3a. Three adsorption peaks associated with the methylene carbon of PEGDE were observed around the chemical shift of 71. The peaks associated with methoxy carbon had a chemical shift around 44.3 and 55.8. To understand the molecular structure of the modified SA fibers in more detail, solid-state 13C-NMR tests were conducted and the results are presented in Figure 3b. For the modified SA fibers, in addition to SA resonance peaks, a new resonance peak around the chemical shift of 71 was observed. This new peak was associated with the methylene carbon of PEGDE. All adsorption peaks shifted due to steric hindrance and hydrogen bonds. Compared with the 13C-NMR spectra of PEGDE in Figure 3a, no peaks with chemical shifts of 40–60 (associated with the methoxy carbon of the epoxy groups) were observed in the modified SA fibers. This indicates that the epoxy groups of PEGDE reacted with the hydroxyl groups of SA and resulted in ether crosslinking. 37
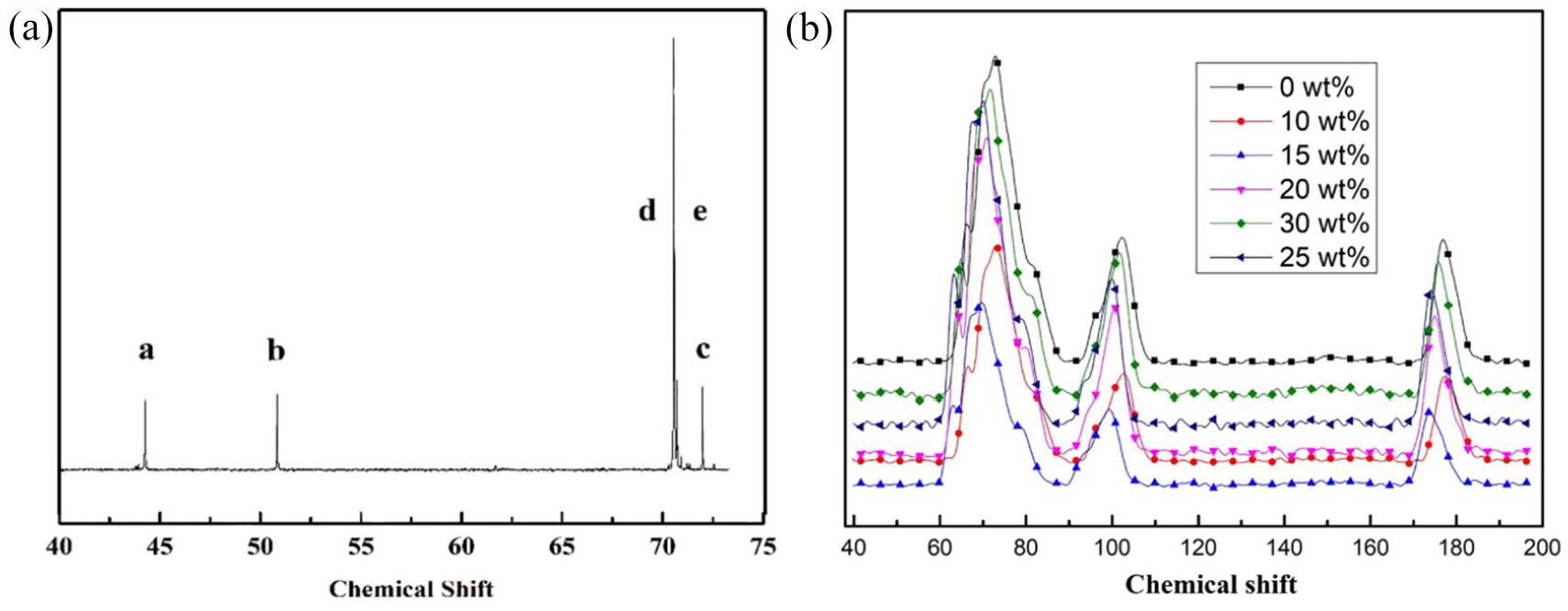
(a) Liquid-state 13C-NMR spectra of PEGDE. (b) Solid-state 13C-NMR spectra of modified SA fibers with various PEGDE contents.
Figures 4 and 5 present the SEM images of the longitudinal surface and cross-sectional surface of modified SA fibers with various PEGDE contents, respectively. It was found that all fibers had different cross-sectional shapes, such as irregular flat and rough circular with non-homogeneous grooves on the surface. The grooves were formed due to the double diffusion between the as-formed fibers and coagulation bath during the spinning process. Such groove structures gave excellent hygroscopicity to the fibers. 40

The longitudinal surface SEM images of modified SA fibers with various PEGDE contents. (a) 0, (b) 10, (c) 15, (d) 20, (e) 25, and (f) 30 wt%.

The cross-sectional SEM images of modified SA fibers with various PEGDE contents. (a) 0, (b) 10, (c) 15, (d) 20, (e) 25, and (f) 30 wt%. The scale bar in the inset images is 5 μm.
Compared with the pure SA fibers, PEGDE-crosslinked fibers had more jagged structures on their surface. This occurred because the carboxyl and hydroxyl groups of the fiber molecules were consumed during the reaction, thereby reducing the fiber hydrophilicity and the water contents. The decreasing fiber hydrophilicity and water contents ultimately introduced more jagged structures to the fiber surface. With the increase of the PEGDE contents, more jagged and stripped structures appeared in the cross sections of modified SA fibers. The “mirror” area in the cross sections of the pure SA fibers disappeared gradually and was replaced by uneven jagged structures.
Figure 6 shows the effects of various PEGDE amounts on the tensile properties of modified SA fibers. As the PEGDE concentration increased, the breaking strength of modified SA fibers increased. When the PEGDE to SA mass ratio reached 15 wt%, the breaking strength of modified SA fibers reached the maximum value of 2.55 cN/dtex, which was 78% higher than the breaking strength of pure SA fibers. The elongation at break of the modified SA fibers was 17.7%, which was about 114% higher than that of the pure SA fibers.
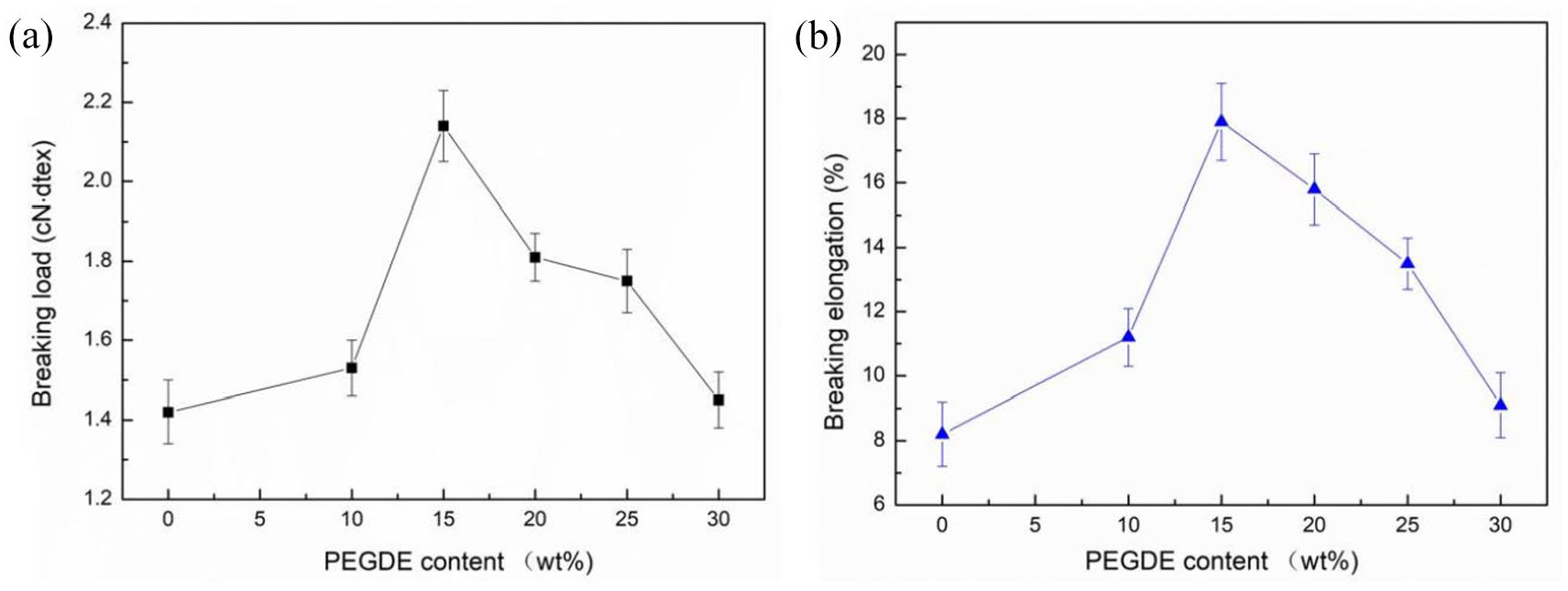
The effect of PEGDE contents on (a) the breaking load and (b) the breaking elongation of modified SA fibers.
PEGDE was crosslinked with SA through the nucleophilic substitution reaction between epoxy end groups in its flexible molecular chains and hydroxyl groups in SA rigid chains. When the PEGDE content was low, the epoxy end groups of the PEGDE molecular chains could easily react with the hydroxyl groups of SA. The number of effective networks in SA fibers increased with the increase in PEGDE contents. However, the grafted PEGDE chains could prevent the latter from grafting on SA chains because the adjacent hydroxyl groups of SA were surrounded by the random coil conformation when the PEGDE content was relatively high (higher than 15 %). Thus, the number of effective networks in SA fibers became lower with the increasing PEGDE contents. 41
Figure 7 presents the thermal properties of modified SA fibers with various PEGDE contents. The thermal degradation of modified SA fibers occurred in four stages. In the first stage (25–230 °C), the fibers lost their free and combined water and hydrogen bonds within, and those among molecules also broke. In the second stage (230–400 °C), glycoside bonds broke and adjacent hydroxyl groups were also removed in the form of water molecules, accompanied by dehydrogenation, decarbonization, and decarboxylation. In the third stage (400–600 °C), intermediate products underwent thermal decomposition and a part of these products underwent carbonization. In the fourth stage (600–800 °C), carbonized products were further decomposed and the remaining volatile products were also removed; consequently, the rate of weight loss slowed. Compared with the pure SA fibers, the maximum thermal degradation temperature of modified SA fibers increased and the corresponding maximum thermal degradation rate decreased. This can be attributed to the increased number of entanglement points in the modified SA fibers. After stretching, the polymer regularity increased, improving the crystallization performance and thermal stability of the modified SA fibers.
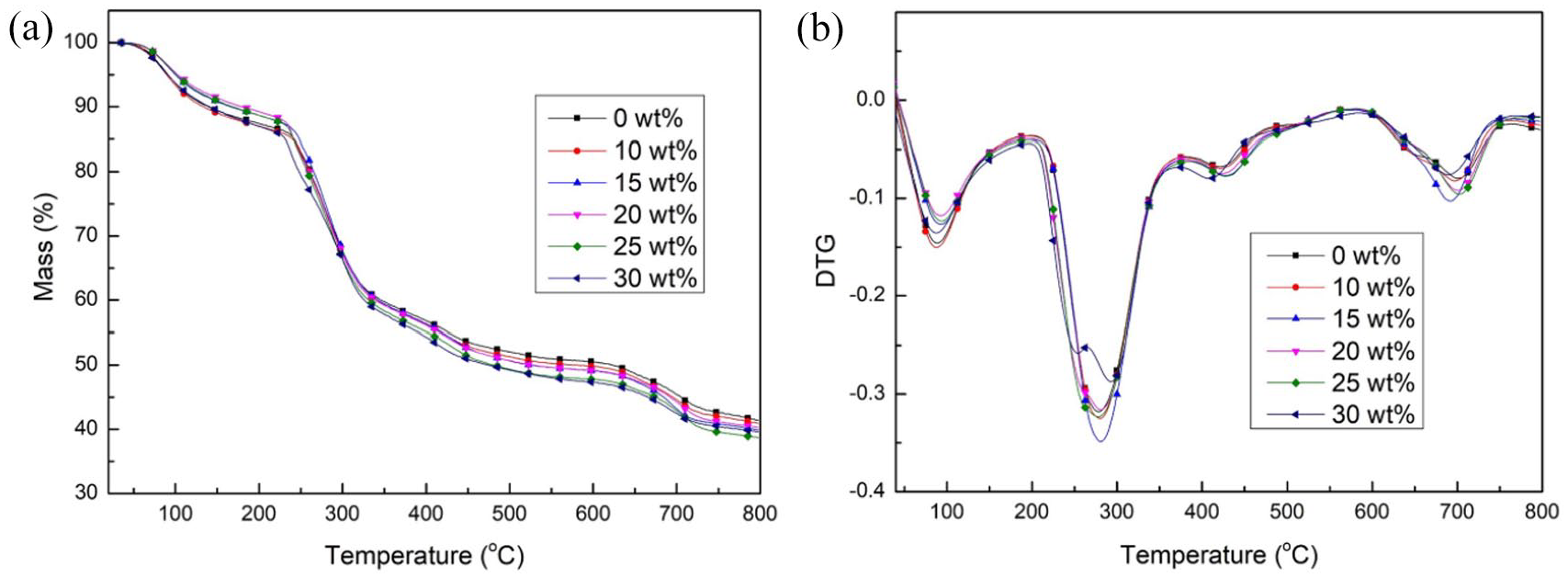
Thermal properties of modified SA fibers with various PEGDE contents. (a) TG and (b) DTG curves.
Conclusion
Modified SA fibers with enhanced mechanical properties can be obtained by spinning the mixed solution of PEGDE and SA after the reaction through a spinneret into a coagulation bath containing aqueous CaCl2. The viscosity of the SA spinning solution increased first and then decreased as the PEGDE contents increased. The structure of the modified SA fibers was characterized and the mechanical properties were measured. PEGDE could crosslink with SA under neutral conditions and subsequently formed a complicated network structure through chelation with Ca2+. Such modification enhanced the breaking strength and elongation at break of modified SA fibers. The optimal breaking strength and the elongation at break were obtained when the PEGDE to SA mass ratio was 15 wt%. Specifically, the breaking strength and elongation at break reached 2.55 cN/dtex and 17.7%, which were 78% and 114% higher than those of the pure SA fibers, respectively. The thermal stability of modified SA fibers was also greatly improved. These novel modified SA fibers have great potential for high-performance clothing and advanced functional wound dressings. The functional SA fibers with excellent mechanical performance will be further investigated in future work.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.