
Abstract
Vitamin B12 deficiency is classically associated with megaloblastic anemia and neurologic or neuropsychiatric manifestations; however, in rare cases, it may present with pancytopenia and laboratory features that mimic hemolytic anemia. Pernicious anemia is a common cause of vitamin B12 deficiency classically described in older adults of Northern European descent; however, it can occur across all ages and ethnic backgrounds, and demographic assumptions should not delay diagnosis. We report a 35-year-old African American man with no significant past medical history who presented with progressive weakness, decreased appetite, and fatigue. Initial laboratory evaluation revealed profound macrocytic anemia, thrombocytopenia, elevated lactate dehydrogenase, indirect hyperbilirubinemia, and low haptoglobin. Despite these findings, the reticulocyte response was inappropriately low, the direct antiglobulin test was negative, and the peripheral blood smear demonstrated no schistocytes, consistent with ineffective erythropoiesis rather than true hemolysis. Further evaluation revealed severe vitamin B12 deficiency with normal folate levels and positive intrinsic factor antibodies, and biopsy findings consistent with autoimmune metaplastic atrophic gastritis supporting a diagnosis of pernicious anemia. Treatment with intramuscular vitamin B12 resulted in rapid clinical and hematologic improvement, emphasizing the reversibility of hematologic manifestations with prompt therapy. This case highlights pernicious anemia as a reversible cause of pseudo-hemolytic anemia and underscores the importance of recognizing its distinguishing features to avoid unnecessary interventions such as plasma exchange or immunosuppressive therapy.
Keywords
Introduction
Vitamin B12, also known as cobalamin, is a water-soluble vitamin obtained primarily from animal-derived foods such as meat, dairy products, and eggs. Its absorption depends on intrinsic factor, a glycoprotein secreted by gastric parietal cells, which binds vitamin B12 and facilitates its uptake in the terminal ileum. 1 After absorption, vitamin B12 serves as an essential cofactor in metabolic pathways involved in DNA synthesis, fatty acid metabolism, and myelin production; consequently, deficiency may manifest with both hematologic abnormalities and neurologic dysfunction. 2 Deficiency of vitamin B12 results in defective DNA synthesis with relative preservation of RNA and protein synthesis, creating a state of nuclear–cytoplasmic asynchrony that underlies the characteristic hematologic manifestations of the disease. 2
Impaired DNA replication in rapidly dividing hematopoietic cells leads to delayed nuclear maturation with continued cytoplasmic growth. In erythroid precursors, this results in macrocytosis, as cells undergo fewer mitotic divisions while continuing to enlarge, producing abnormally large red blood cells with an elevated mean corpuscular volume (MCV). 3 Similar defects in nuclear maturation affect myeloid precursors, giving rise to hypersegmented neutrophils, often the earliest and most sensitive peripheral blood finding of megaloblastic anemia. 4 In more severe cases, defective DNA synthesis triggers cell-cycle arrest and apoptosis of erythroid precursors within the bone marrow, a process termed ineffective erythropoiesis. 5 This intramedullary destruction of hematopoietic cells leads to cytopenias, an inappropriately low reticulocyte response, and the release of intracellular contents such as lactate dehydrogenase and bilirubin, producing laboratory features that can closely mimic hemolytic anemia despite the absence of true peripheral red blood cell destruction.5,6
In the general population, vitamin B12 deficiency accounts for approximately 1–2% of anemia cases. 6 Although historically associated with older age and limited dietary intake, vitamin B12 deficiency remains common across diverse populations, affecting approximately 6% of adults younger than 60 years and up to 20% of adults older than 60 years in the United States. 6 The most common etiologies include nutritional deficiency, malabsorption syndromes, and autoimmune destruction of gastric parietal cells leading to pernicious anemia. 5 Pernicious anemia represents the most frequent cause of severe vitamin B12 deficiency in developed countries, with an estimated prevalence of approximately 0.1% in the general population and nearly 2% among adults over 60 years.5,6
Pernicious anemia arises from autoimmune atrophic gastritis mediated by autoantibodies directed against intrinsic factor and the gastric parietal cell H+/K+ adenosine triphosphatase, resulting in achlorhydria, intrinsic factor deficiency, and impaired cobalamin absorption.5,7 It frequently coexists with other autoimmune disorders, including autoimmune thyroid disease, type 1 diabetes mellitus, vitiligo, and Addison’s disease, reflecting a shared predisposition to immune dysregulation.8,9 Progressive immune-mediated injury exposes additional gastric antigens through epitope spreading, ultimately resulting in parietal cell loss, reduced intrinsic factor production, and impaired vitamin B12 absorption. 9
In rare cases, advanced vitamin B12 deficiency presents with pancytopenia and laboratory features suggestive of hemolytic anemia, including markedly elevated lactate dehydrogenase, indirect hyperbilirubinemia, and low haptoglobin.7,8 This pseudo–hemolytic or thrombotic microangiopathy–like picture reflects ineffective erythropoiesis with intramedullary destruction of hematopoietic precursors rather than true hemolysis or microvascular thrombosis.7,8 Here, we describe the case of a previously healthy young adult who presented with severe bicytopenia and hemolysis-like laboratory abnormalities secondary to pernicious anemia, highlighting vitamin B12 deficiency as an important and reversible mimic of hemolytic disorders.
Case Presentation
A 35-year-old African American male with no significant past medical history presented to the Emergency Department with one week of progressive generalized weakness, associated with fatigue, nausea, and vomiting.
On admission, vital signs were notable for a temperature of 98°F, blood pressure 108/75 mmHg, heart rate 87 beats per minute, respiratory rate 19 breaths per minute, and oxygen saturation 97% on room air. Cardiovascular examination demonstrated normal S1 and S2 without murmurs. Lungs were clear to auscultation bilaterally. Abdominal examination showed a soft, nondistended, and nontender abdomen without hepatosplenomegaly. No peripheral edema was present, and neurologic examination was nonfocal, with no impairment of proprioception or cognition noted. No scleral icterus or pallor was noted.
Relevant Laboratory Workup
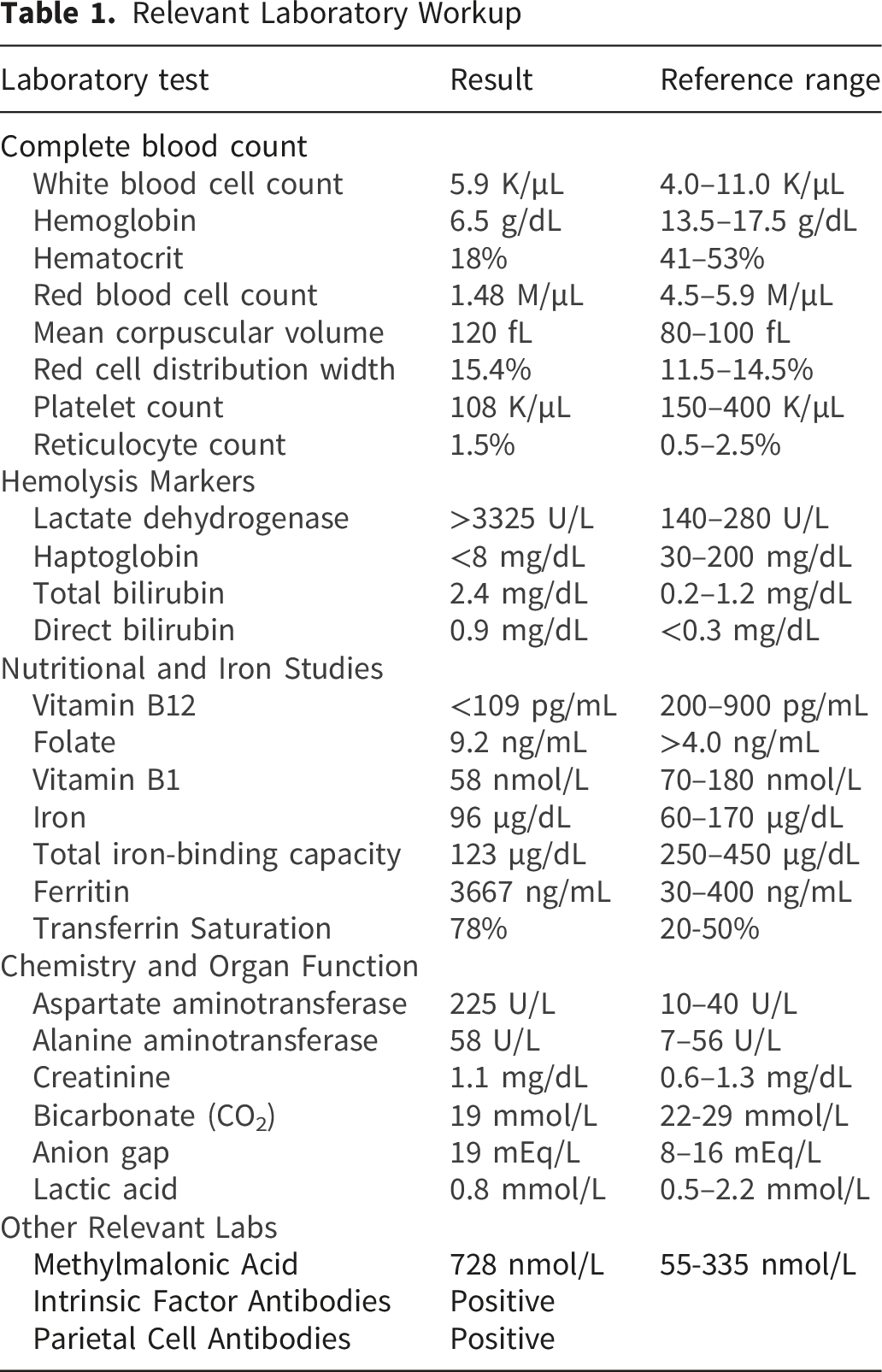
Further testing revealed severe vitamin B12 deficiency (<109 pg/mL) with normal folate levels. Methylmalonic acid and homocysteine levels were elevated, confirming functional vitamin B12 deficiency. Iron studies demonstrated markedly elevated ferritin (3,667 ng/mL) with low total iron-binding capacity.
Autoimmune evaluation revealed positive intrinsic factor blocking antibodies and parietal cell IgG antibodies, confirming pernicious anemia as the underlying etiology. GI was consulted at this time for endoscopic evaluation.
Upper endoscopy demonstrated histology consistent chronic autoimmune metaplastic atrophic gastritis on biopsy taken from the gastric body (Figures 1 and 2), while colonoscopy revealed only hyperplastic colonic polyps. No active source of gastrointestinal bleeding was identified. Gastric body biopsy: Microscopic examination demonstrating loss of parietal cells in a background of lymphoplasmacytic infiltrate in the lamina propria. Blue arrow: lymphocytic infiltrate. Orange arrow: intestinal metaplasia. Gastric body biopsy: Microscopic examination demonstrating diffuse lymphoplasmocytic infiltrate. Red arrow: destruction of sparsely seen parietal cells.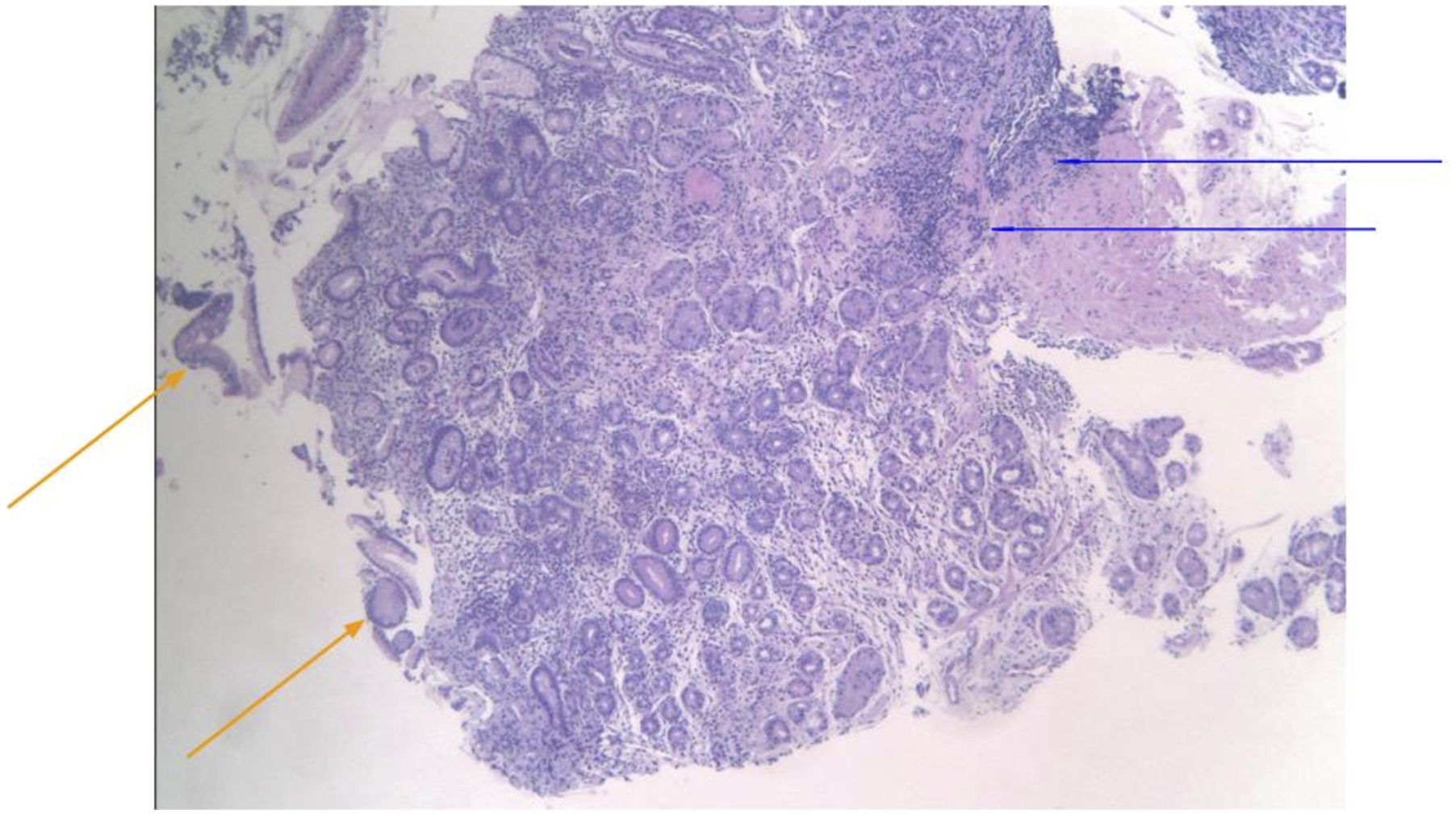

Alternative etiologies for the patient’s cytopenias and hemolysis-like laboratory findings were systematically evaluated and ruled out. Thrombotic microangiopathy, including thrombotic thrombocytopenic purpura, was felt to be unlikely given the absence of schistocytes on multiple peripheral blood smears, preserved renal function, and lack of neurologic manifestations. Autoimmune hemolytic anemia was excluded by a negative direct antiglobulin test (DAT), and disseminated intravascular coagulation was ruled out based on normal coagulation parameters and stable fibrinogen levels. Although DAT negative hemolysis and bicytopenia in a relatively young patient may raise concern for paroxysmal nocturnal hemoglobinuria (PNH), this diagnosis was considered unlikely and flow cytometry was not performed due to a number of factors, including reticulocytopenia, where reticulocytosis would be expected in PNH, elevated MCV atypical of isolated PNH, lack of clinical symptoms such as dark urine, and more compelling evidence favoring a diagnosis of vitamin B12 deficiency. There was no history of heparin exposure to suggest heparin-induced thrombocytopenia, and mechanical hemolysis was excluded by the absence of schistocytes or spur cells on peripheral smear. Primary bone marrow disorders, including aplastic anemia, myelodysplastic syndrome, and acute leukemia were considered; however, these were deemed unlikely given the marked macrocytosis, biochemical evidence of vitamin B12 deficiency, lack of dysplasia or blasts on peripheral smear, and subsequent hematologic improvement following vitamin B12 replacement. Although iron studies demonstrated markedly elevated ferritin and transferrin saturation, these findings were attributed to ineffective erythropoiesis and increased cellular turnover rather than primary iron overload; hereditary hemochromatosis was effectively excluded after high iron Fe gene (HFE) genetic testing demonstrated heterozygosity without evidence of clinically significant iron overload. Overall, the constellation of cytopenias, markedly elevated lactate dehydrogenase, indirect hyperbilirubinemia, low haptoglobin, and an inappropriately low reticulocyte response was most consistent with ineffective erythropoiesis and intramedullary red blood cell destruction due to severe vitamin B12 deficiency rather than true peripheral hemolysis.
Liver enzymes demonstrated aspartate aminotransferase (AST)–predominant transaminitis (AST 225 U/L, alanine aminotransferase (ALT) 58 U/L with hyperbilirubinemia, normal alkaline phosphatase, and preserved hepatic synthetic function. An evaluation for alternative causes of hepatitis was pursued, and viral hepatitis was excluded by a nonreactive hepatitis panel and absence of an acute viral prodrome, while autoimmune hepatitis was ruled out based on negative antinuclear and anti–smooth muscle antibodies, normal immunoglobulin levels, and clinical improvement without immunosuppressive therapy. Wilson disease was also considered and excluded by normal ceruloplasmin levels. Ultimately, the liver enzyme abnormalities were attributed to ischemic and hemolysis-related hepatic injury in the setting of severe anemia and intramedullary hemolysis, supported by AST predominance and improvement following vitamin B12 repletion.
Given the severity of anemia, the patient received packed red blood cell transfusion on admission with appropriate clinical and laboratory response. The patient was initiated on parenteral vitamin B12 replacement with intramuscular cyanocobalamin 1,000 µg daily, consistent with recommended therapy for severe deficiency with symptomatic anemia. With treatment, hemoglobin stabilized, MCV began to downtrend, and bilirubin and transaminases improved. Following hematologic stabilization and in the absence of neurologic deficits, the patient was transitioned to high-dose oral cyanocobalamin 1,000 µg daily with plans for close outpatient monitoring. Although intrinsic factor deficiency in pernicious anemia impairs conventional enteral absorption, high-dose oral therapy remains effective via passive diffusion independent of intrinsic factor, with absorption of approximately 1% of the dose administered - sufficient to maintain cobalamin repletion in adherent patients.
The patient was discharged in stable condition with planned outpatient follow-up with hematology to monitor hematologic response to vitamin B12 replacement and with gastroenterology for repeat endoscopic surveillance given the diagnosis of autoimmune metaplastic atrophic gastritis and associated malignancy risk.
Discussion
This case highlights a unique presentation of severe vitamin B12 deficiency caused by pernicious anemia, with initial lab findings of bicytopenia with biochemical markers of hemolysis. The primary diagnostic challenge presented in this case was pseudo-hemolysis - intramedullary destruction of erythroid precursors producing a biochemical profile that closely mimicked peripheral red blood cell destruction. This presentation may closely mimic primary hemolytic disorders, including primary hemolytic anemia or thrombotic microangiopathy (TMA), due to the aforementioned processes of intramedullary hemolysis and ineffective hematopoiesis. However, this presentation is quite rare, as only 10% of patients with vitamin B12 deficiency will present as this patient did, with symptomatic anemia, pancytopenia, and hemolysis.10,11
The biochemical profile demonstrated in this case of elevated LDH, indirect hyperbilirubinemia, and undetectable haptoglobin illustrates the release of intracellular contents of erythroid precursors as a result of intramedullary destruction. This process can be further localized to the bone marrow, and distinguished from peripheral hemolysis, by considering the lack of schistocytes and inappropriately low reticulocyte response, suggestive of impaired DNA synthesis in the setting of vitamin B12 deficiency. As previously described, vitamin B12 serves as an essential element in the synthesis of DNA as a cofactor for methylcobalamin in the Methionine synthase pathway, and is necessary for cellular metabolism, DNA synthesis, and red blood cell development. 12
While the core diagnostic challenge in this case was pseudo-hemolysis, it may be worth noting that severe vitamin B12 deficiency may also present with the presence of schistocytes on peripheral smear, further obscuring the diagnosis towards a potential TMA, or pseudo-TMA, in roughly 2.5% of cases. 13 In this case, in the absence of schistocytes and presence of preserved renal function and mild thrombocytopenia, both true and pseudo TMA were excluded. However, the rarity of this phenomenon underscores the importance of prompt recognition, as misdiagnosis may subject patients to unwarranted interventions such as unnecessary plasma therapy, immunosuppression or bone marrow biopsy.
Differentiating pseudo-TMA secondary to vitamin B12 deficiency from true TMAs such as thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS) is critical. Reticulocytopenia has been described as the most reliable indicator separating B12 deficiency from true TMA, 14 with reticulocyte counts less than 3% highly suggestive of vitamin B12 deficiency. Marked elevations in LDH have also been described and should be considered. In one systematic review including 41 patients with vitamin B12 deficient pseudo-TMA the median LDH was 3,981 U/L, with half of the patients falling between 2,004 and 5,467 U/L. 14 By comparison, the Oklahoma TTP registry from 1989-2008 reported a median LDH of 1,090 U/L (interquartile range 114-12,587) among 201 patients with A Disintegrin and Metalloproteinase with Thrombospondin type-1 motif, member 13 (ADAMTS13) levels greater than 10%, and 1,407 U/L (interquartile range 256-3,909) among 60 patients with ADAMTS13 levels less than 10%. 15 The median LDH in pseudo TMA exceeds that reported in TTPm although overlap between the ranges is considerable. In this case, the LDH exceeded 3,325 U/L, the upper limit of the laboratory assay’s detection range, consistent with the markedly elevated values characteristically seen in pseudo-TMA. Both reticulocytopenia and markedly elevated LDH were present in this case, and early and accurate recognition of pseudo-TMA is critical in order to avoid unnecessary plasma therapy and to prompt appropriate treatment with vitamin B12 replacement.
Unfortunately, misdiagnoses surrounding vitamin B12 deficiency and pseudo-TMA are not uncommon, and a literature review by Tun et al noted that nearly 40% of cases of pseudo TMA caused by vitamin B12 deficiency were initially diagnosed as TTP and treated with plasma therapy. 16 As opposed to the low cost, efficient, and tolerable treatment of B12 deficiency with supplementation, the misdiagnosis of TTP carries additional risks associated with plasma exchange, ICU admission, and exposure to blood products.
In adults, pernicious anemia is the most common cause of vitamin B12 deficiency and accounts for up to 50% of cases. Characteristics of pernicious anemia include the presence of intrinsic factor antibodies, parietal cell antibodies, fundic atrophy, and destruction of the gastric mucosa via cell mediated autoimmunity. 7 However, dietary causes of vitamin B12 deficiency must not be overlooked, as an age dependent incidence is present in the elderly community, with rates of deficiency ranging from 12-40%, 17 and other studies noting deficiency by population to be up to 90% in the elderly and 86% among children, with higher rates among vegetarians and vegans. 18 It is also worth noting that pernicious anemia is classically described as a disease of older adults of Northern European descent; however, this case illustrates that the diagnosis can occur across all age groups and backgrounds, and this patient’s profile as a young African American male should serve as a reminder that demographic assumptions should not be used to dismiss diagnoses. Identifying the etiology of B12 deficiency, and determining whether it is due to diet or autoimmune dysfunction, has clear clinical implications in lifelong management.
Pernicious anemia presents as a late stage of autoimmune gastritis, which is often present with parietal cell antibodies and intrinsic factor antibodies, as in this patient. Furthermore, histopathologic features suggestive of autoimmune gastritis include lymphoplasmacytic infiltrate in the basal lamina propria and significant loss of oxyntic glands, 19 which can be seen on microscopic examination of a biopsy taken from a pseudo polyp in the body of this patient’s stomach. Furthermore, with the presence of intestinal metaplasia, the clinical picture and pattern seen on biopsy are suggestive of autoimmune atrophic gastritis.
It is therefore imperative to keep B12 deficiency pseudo TMA in mind when treating a patient presenting with hemolysis. This case serves to add to the existing body of literature and highlight key distinguishing factors for clinicians to look for that can differentiate the types of hemolysis. In the case of pseudo-TMA secondary to vitamin B12 deficiency, key lab results to note on routine workup include reticulocytopenia, marked elevation of LDH, and peripheral smear interpretation. This case also serves to add to the limited number of described cases of adults with pancytopenia and hemolysis secondary to cobalamin deficiency, with the aim of decreasing the incidence of misdiagnosis and subsequent mistreatment.
Conclusion
Pseudo-hemolytic anemia secondary to vitamin B12 deficiency is a rare phenomenon and creates a diagnostic challenge. The pseudo hemolytic presentation may mimic other forms of hemolysis by levels of LDH, haptoglobin, indirect hyperbilirubinemia, anemia, symptoms, and physical exam findings. However, key clues that warrant consideration of B12 deficiency include macrocytosis, markedly elevated LDH, and reticulocytopenia, which are often available on initial workup. Prompt recognition and appropriate treatment with supplementation often results in rapid reversal of abnormalities and improvement in symptoms. When unrecognized, however, patients are subjected to unwarranted therapies for TMA, such as plasma exchange therapy and ICU admission. The purpose of this case report is to increase provider awareness of a relatively rare presentation, avoid unnecessary morbidity, and facilitate prompt diagnosis and appropriate management.
Footnotes
Acknowledgments
We would like to express our gratitude to the clinical staff at The Brooklyn Hospital Center for their exceptional care and support in the management of the patient described in this case report. We also thank our colleagues for their insightful discussions and contributions to the development of this manuscript. Lastly, we appreciate the guidance and resources provided by the Institutional Review Board, which facilitated ethical approval for publication.
Ethical Considerations
Permission to report this case was granted by the Institutional Review Board of The Brooklyn Hospital Center (IRB No. 2263278).
Consent to Participate
Written informed consent was obtained from the patient for publication of their anonymized clinical information in this article.
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Prior Presentation of Abstract
This abstract has not been previously presented at any meeting or conference.