
Abstract
Spastic tetraplegia, thin corpus callosum, and progressive microcephaly (SPATCCM) is a rare autosomal recessive neurodevelopmental disorder caused by pathogenic variants in the SLC1A4 gene. 24 cases have been reported worldwide. We describe a 30-month-old boy born to consanguineous parents, presenting with global developmental delay, seizures, progressive microcephaly, spastic tetraplegia, feeding difficulties, and recurrent infections. Genetic testing confirmed a homozygous pathogenic variant in SLC1A4 (p.Arg457Trp). Notably, our patient exhibited novel systemic phenotypic features, including grade 2 finger clubbing, recurrent infections, bilateral hernias, meatal stenosis, and dysmorphic features (high-arched palate, low-set ears, pectus carinatum). Brain MRI demonstrated a diffusely thin corpus callosum, a typical finding consistent with the classic SPATCCM phenotype. These previously unreported systemic features broaden the recognized phenotypic spectrum and underscore the need for genetic testing in suspected cases.
Keywords
Introduction
The SLC1A4 gene encodes the alanine–serine–cysteine transporter 1 (ASCT1), a sodium-dependent neutral amino acid transporter that mediates the transfer of amino acids, particularly L-serine, from astrocytes to neurons. This process is essential for normal brain development and neuronal function. 1 Pathogenic variants in SLC1A4 cause a very rare autosomal recessive neurodevelopmental disorder known as spastic tetraplegia, thin corpus callosum, and progressive microcephaly (SPATCCM). 2 SPATCCM is characterized by early-onset spastic tetraplegia, progressive microcephaly, thinning of the corpus callosum, and severe global developmental delay beginning in infancy. The disorder was first described simultaneously in 2015 by Srour et al, who reported two affected siblings from an Ashkenazi Jewish consanguineous family, 3 and Damseh et al, who described 11 patients (10 Ashkenazi Jewish and 1 Palestinian). 4 To date, 24 affected individuals have been reported worldwide, with an estimated prevalence of less than 1 in 1,000,000. 5 Subsequent cases have been described in several other ethnic backgrounds, including Irish, Italian, Czech, Palestinian, and Pakistani populations. As demonstrated across reported cases, diagnosis has involved a combination of characteristic clinical features, brain magnetic resonance imaging findings, and molecular confirmation of pathogenic variants in SLC1A4, most commonly identified using whole-exome sequencing.1,4 Therapeutic approaches using L-serine supplementation have been attempted in disorders of serine biosynthesis; however, the benefit of serine supplementation in serine transport defects remains uncertain, as the major neurological abnormalities are thought to arise during early intrauterine development. 2 Severe neurological impairment in classical serine deficiency disorders can only be effectively prevented through prenatal or very early postnatal administration of L-serine, a limitation that should be considered when evaluating serine replacement therapy in SPATCCM. 6 Here, we report a child with a pathogenic variant in SLC1A4 who demonstrates clinical features that partially overlap with previously reported cases, while also exhibiting novel manifestations that further expand the phenotypic spectrum of this ultra-rare disorder.
Case Presentation
The patient is a 30-month-old male, born at 39 weeks of gestation to consanguineous parents (first cousins), with a birth weight of 1950 grams and a head circumference (HC) of 28 cm. He presented to our clinic with a history of progressive microcephaly, spastic tetraplegia, recurrent infections, and dysphagia, all of which were evident since early infancy. His motor development has been significantly delayed, as he was unable to sit independently or roll over by 8 months of age. While he showed weight-bearing in his lower extremities, he lacked purposeful upper extremity reach. Social and communication development was also delayed, though he maintained preserved eye contact.
His medical and surgical history includes the repair of bilateral inguinal and umbilical hernias at 2 months of age and a meatal stenosis repair at 9 months. Seizures began at 4 months of age and were managed with sodium valproate, with carbamazepine added at 26 months of age, while baclofen was used for spasticity control. Despite these interventions, he has experienced frequent recurrent infections (approximately every 10 days) since birth. His feeding difficulties, noted since birth, were further evaluated with a video fluoroscopy swallowing study, which revealed impaired bolus control and inadequate management of textured food, especially thicker consistencies. Due to his compromised oral musculature, a G-tube placement was recommended for long-term nutritional support.
On physical examination, performed at 30 months of age, the patient’s HC measured 38 cm, indicating continued microcephaly. His growth parameters included a weight of 7 kg and a length of 73 cm. Head circumference-for-age demonstrated severe progressive microcephaly, with values consistently below the 3rd percentile (Z-score ≈ −7.8 at 30 months). Weight-for-age and length-for-age were markedly below the expected range throughout follow-up, corresponding to severe weight-for-growth restriction (Z-score ≈ −4.7) and severe length-for-age growth restriction (Z-score ≈ −5.3) at 30 months of age. Craniofacial assessment was significant for microcephaly, retrognathia, and a high-arched palate (Figure 1A and B). The ears were noted to be low-set and posteriorly rotated with a prominent antihelix (Figure 1C). Clinical photographs of the patient at 30 months.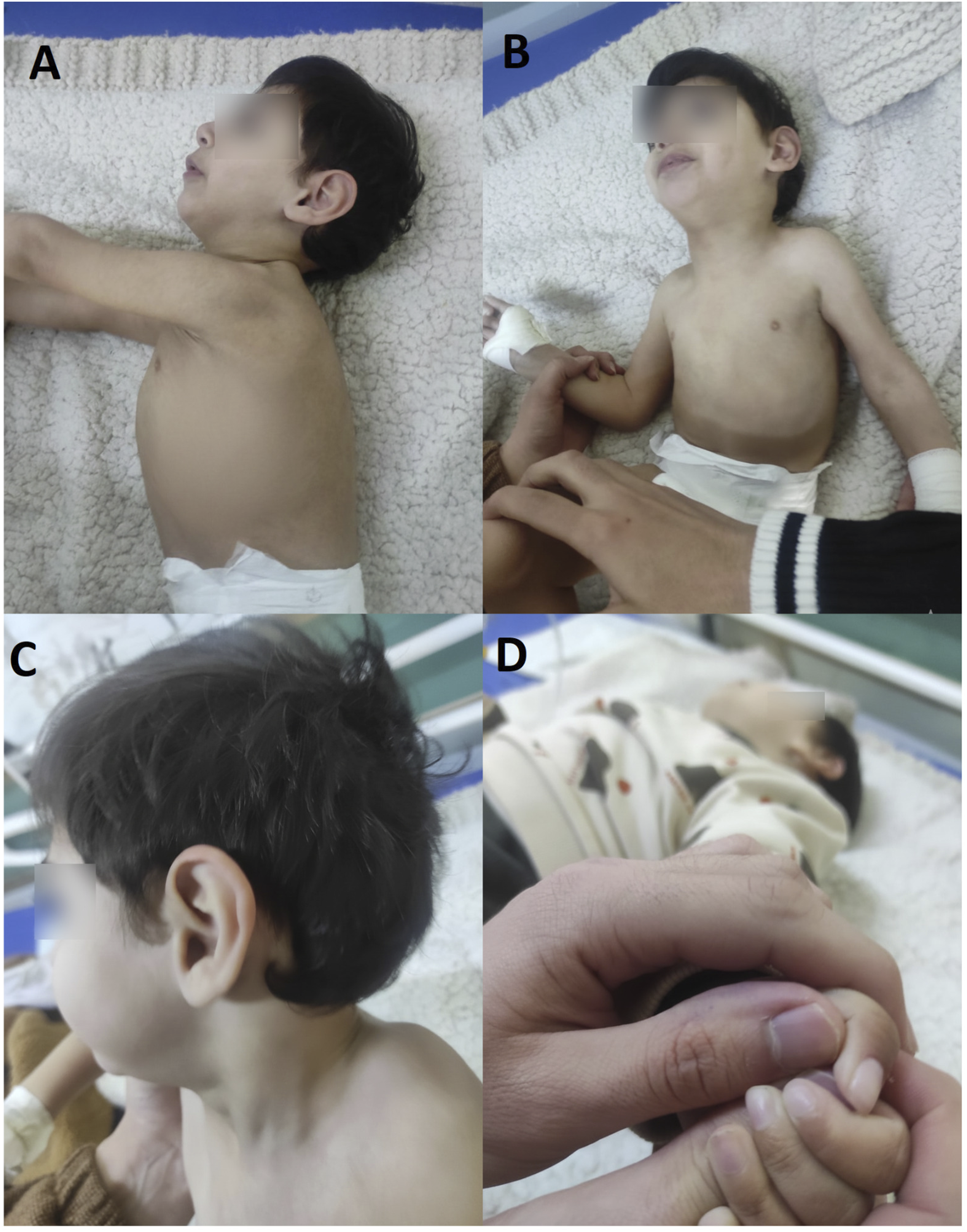
The patient’s nutritional status was consistent with failure to thrive, evidenced by a lack of subcutaneous fat and visible rib prominence. Thoracic examination revealed a distinct pectus carinatum (Figure 1A). Neurologically, the patient demonstrated a combination of axial hypotonia and limb spasticity consistent with spastic tetraplegia and global developmental delay. Examination of the extremities revealed grade 2 digital clubbing, characterized by the softening of the nail beds and loss of the Lovibond angle (Figure 1D). Brain imaging, including MRI performed at 7 months of age, was negative for acute infarct, hemorrhage, or ventriculomegaly. However, imaging revealed a diffusely thin corpus callosum despite the presence of all anatomical segments (rostrum, genu, body, and splenium), suggesting a primary structural hypoplasia rather than an acquired injury (Figure 2). An auditory examination revealed normal tympanometry, DPOAE, and ABR responses, indicating no hearing loss. Neuroimaging at 7 months of age.
The patient was diagnosed with spastic tetraplegia and progressive microcephaly, which was later confirmed through whole-exome sequencing. Genetic testing identified a homozygous pathogenic variant in SLC1A4 (NM_003038.5:c.1369C>T, p.Arg457Trp), diagnosing him with an autosomal recessive neurodevelopmental disorder known as SPATCCM. His family history is notable for the consanguinity of his parents. The parents are clinically unaffected; however, parental genotyping was not available. Given the autosomal recessive inheritance pattern of SLC1A4-related SPATCCM and the consanguinity of the parents, heterozygous carrier status is strongly inferred.
The patient’s older sibling, a 3-year-old female, demonstrated normal head circumference (48 cm, 50th percentile) and age-appropriate developmental milestones. Subsequent testing confirmed she is heterozygous for the SLC1A4 (NM_003038.5:c.1369C>T; p.Arg457Trp) pathogenic variant, consistent with carrier status. Genetic counseling was provided to the family regarding autosomal recessive inheritance and future reproductive options. Whole-exome sequencing (WES) was performed at Hadassah Hospital (Jerusalem) using the Illumina NovaSeq platform (average coverage >100×; 98% ≥20×). Bioinformatic analysis followed ACMG 2021 guidelines.
WES identified a homozygous SLC1A4 variant (NM_003038.5:c.1369C>T; p.Arg457Trp). This variant is classified as pathogenic according to ACMG criteria and is annotated as pathogenic/likely pathogenic in ClinVar. It has been previously reported in individuals with spastic tetraplegia, thin corpus callosum, and progressive microcephaly. The variant occurs at a very low allele frequency in population databases (∼0.00012), with no healthy homozygotes reported, further supporting its pathogenicity in autosomal recessive SLC1A4-related disease.
No chromosomal microarray or additional genetic testing was performed. Whole-exome sequencing does not capture noncoding regions, genes with high homology to pseudogenes, or repeat expansions such as Fragile X. CNV and SV analysis did not reveal any relevant findings. Therefore, although technical limitations prevent the absolute exclusion of all possible genetic contributors, no evidence of an alternate primary or secondary diagnosis was identified.
Discussion
SLC1A4 was identified as a disease-causing gene in 2015 by Srour et al, 3 who described biallelic mutations in an Ashkenazi Jewish family, and Damseh et al, 4 who described biallelic mutations in Ashkenazi Jewish and Palestinian families, naming the condition SPATCCM. Later studies reported novel pathogenic variants in patients from Pakistani, 2 Portuguese, 7 and Turkish populations. 5
SLC1A4-related disorders result from biallelic pathogenic variants in the SLC1A4 gene, which encodes the ASCT1 serine transporter.4,8 Parental consanguinity has been reported in families with SLC1A4-related disorder, consistent with its autosomal recessive inheritance pattern.3,4 Phenotypic severity correlates with mutation type in SLC1A4, with combined loss-of-function and missense variants resulting in more severe disease than missense variants alone. 8 In serine biosynthesis defects, a clinical spectrum ranging from severe infantile to milder juvenile phenotypes has been described, though biochemical findings cannot reliably predict clinical severity.9,10 Serine supplementation as a therapeutic approach in SLC1A4 disorder remains hypothetical. 8
SLC1A4 pathogenic variants impair the ASCT1 transporter, reducing L-serine uptake in the brain.4,8,11 L-serine is essential for neuronal proliferation, migration, myelination, and synthesis of phospholipids and D-serine, the latter being a co-agonist of NMDA receptors.10,12 Deficiency of L-serine disrupts these processes, contributing to the neurological features of SPATCCM.
SLC1A4-related disorders present with three defining neurological features: (1) progressive microcephaly beginning in infancy, (2) spastic tetraplegia with hypertonia, and (3) a thin corpus callosum, often with hypomyelination visible on brain imaging.3,4,8 Affected individuals commonly show global developmental delay and intellectual disability ranging from moderate to severe.3,4,8 Seizures are a recognized feature of SLC1A4-related disorder, with manifestations ranging from simple febrile seizures to intractable epilepsy.3,4,8,13 Additional features include feeding difficulties, growth retardation, and occasionally stereotypic hand movements.4,8,9
In our case, the patient exhibited the classic phenotype of SPATCCM, including progressive microcephaly, severe global developmental delay, spastic tetraplegia, and seizures. Dysphagia was confirmed by videofluoroscopy, and persistent low weight and length were noted. A significant communication delay was observed, as the patient produced only babbling without meaningful speech, while auditory examination excluded hearing impairment. Notably, his older sister was found to be heterozygous for the same variant and demonstrated normal development, consistent with carrier status. However, objective clinical examination, neuroimaging, and formal neurodevelopmental testing were not available.
Comparison of Phenotypic Findings and SLC1A4 Variants Across Reported Cases

✓ Present, ✗ Absent, NR = Not Reported.
Notes. NR = Not Reported. Frequency estimates are based on all published SLC1A4 cases as of 2024. Recurrent infections, Digital clubbing, genitourinary anomalies, and hernias represent novel features first documented in the current case.
The diagnosis of SPATCCM is suspected based on a combination of characteristic clinical features and confirmed by genetic testing.1,4 Key diagnostic criteria include early infantile onset of severe neurodevelopmental delay, progressive microcephaly, and spastic tetraplegia.2,4 Brain imaging findings such as thin corpus callosum, hypomyelination, and white matter abnormalities are frequently observed and considered a hallmark of the condition, but they are not universally present.1,2,4 In our patient, neuroimaging at 7 months of age demonstrated a diffusely thin corpus callosum, consistent with the classic SPATCCM phenotype. Mid-sagittal T2-weighted sequences confirmed that while the rostrum, genu, body, and splenium are present, they lack the expected age-appropriate thickness and bulbous morphology.
Notably, other imaging abnormalities often associated with this condition were absent; coronal T1-weighted images showed normal symmetric brain development with no evidence of ventriculomegaly, cortical volume loss, or gross delays in myelination. The preservation of the brainstem and cerebellar vermis further highlights the variability in structural brain changes, suggesting that white matter abnormalities may be a downstream or non-universal consequence of the underlying serine transport defect. These findings underscore the importance of proceeding to genetic testing for a definitive diagnosis, even when global brain structure appears otherwise unremarkable.
Management is primarily supportive, with an emphasis on seizure control. 15 Serine supplementation has been proposed as a potential targeted therapy, given the overlapping features with L-serine biosynthesis disorders in which supplementation appeared beneficial; however, its efficacy in SLC1A4 deficiency has not been established.4,15 In our patient, seizures began at 4 months of age and were managed with sodium valproate, with carbamazepine added at 26 months of age, while baclofen was used for spasticity control.
Conclusion
This case expands the phenotypic spectrum of SLC1A4-related SPATCCM by documenting previously unreported or rarely described multisystem manifestations, including genitourinary anomalies, recurrent infections, and digital clubbing. Neuroimaging demonstrated a diffusely thin corpus callosum, a hallmark feature of SPATCCM, in the absence of additional major structural brain abnormalities. These findings highlight the clinical variability of SLC1A4-related disorders and underscore the importance of molecular genetic testing for definitive diagnosis, even when neuroimaging does not demonstrate extensive white matter involvement. Recognition of such phenotypic variability enhances clinical awareness, supports accurate genetic counseling, and may inform future therapeutic considerations, including early targeted metabolic interventions, whose efficacy in this disorder remains to be established.
Footnotes
Acknowledgements
The authors express their gratitude to the patient and their family for their great contribution. Also the authors express profound gratitude to the Polytechnic Medical Students’ Research Association (PMRA) for their invaluable contributions and unwavering support that significantly enriched every stage of the research journey.
Ethical Considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Consent for Publication
Written informed consent was obtained from the patient’s family for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal if requested.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data used to support the findings of this study are included in the article.