
Abstract
Primary intestinal lymphangiectasia (PIL) is a rare protein-losing enteropathy, typically diagnosed in childhood. Adult-onset PIL is exceptionally rare and poses a significant diagnostic challenge, often diagnosed as other gastrointestinal diseases. A 28-year-old female, presented with recurrent chylous ascites, and hypoalbuminemia (2.3 g/dL); no hepatic dysfunction or immunoglobulin deficiency was evident; comprehensive evaluation excluded hepatic, malignant (lymphoma), and infectious etiologies (tuberculosis, filariasis). Past history is significant for a presumptive diagnosis of Crohn’s disease, which was initially made based on clinical picture and laboratory finding of elevated fecal calprotectin, however, endoscopy and histology studies were inconclusive. On further investigations, imaging showed colonic wall thickening with mesenteric lymphadenopathy, and ascitic fluid analysis showed chylous ascites without evidence for malignant or infectious etiology. Upper endoscopy revealed multiple white duodenal plaques, and terminal ileal biopsies confirmed notable lymphatic dilation. Treatment was initiated with dietary modification (low-fat, high-protein, medium-chain triglyceride supplementation) and budesonide; the patient showed partial response. Later on, Octreotide therapy was initiated, and led to gradual resolution of ascites. Adult onset PIL is challenging to diagnose, particularly when initially misdiagnosed as an inflammatory bowel disease. For correct diagnosis, thorough evaluation, by histopathology and exclusion of secondary causes, is essential. Dietary therapy is the mainstay of management; additional benefits can be obtained by pharmacologic options like octreotide in refractory cases. This case is among the first reported cases of adult-onset PIL from Palestine, contributing to the limited literature and highlighting the need for heightened clinical awareness of such rare presentations.
Keywords
Introduction
Primary intestinal lymphangiectasia (PIL) is defined as a rare congenital protein losing enteropathy, caused by dilated intestinal lymphatics resulting in lymph leak into the gastrointestinal tract, its other name is Waldmann’s disease. 1
PIL is mainly diagnosed in children, often before the age of three, and few reports exist regarding adult onset PIL, which highlights how PIL is poorly understood, often misdiagnosed, and needs further investigation. The clinical presentation is highly variable, ranging from asymptomatic forms to severe protein-losing enteropathy, which contributes to the uncertainty of its true prevalence.2,3
Symptomatic patients complain of diarrhea, abdominal pain, variable degrees of peripheral edema or serous effusions due to lymphatic drainage impairment and protein loss, signs of malabsorption, and their laboratory studies reveal hypoalbuminemia, lymphopenia, hypogammaglobulinemia.1,4
Misdiagnoses or diagnostic delay often happens, with many reasons explaining the rarity of the condition; its overlapping symptoms with other more common diseases like inflammatory bowel disease or celiac disease, in addition to the absence of a known protocol or recognizable diagnostic roadmap in adults.5,6
In addition to thorough clinical assessment, diagnosis requires laboratory, imaging and histopathological studies, exclusion of secondary causes (e.g: cardiac disease, lymphoma, infections, portal hypertension) of intestinal lymphangiectasia (IL) remains a cornerstone in diagnosis.1,5
Advances in diagnostic modalities, including capsule endoscopy, push-or double-balloon enteroscopy, radiologic lymphatic imaging (e.g., MR lymphangiography), and lymphatic embolization, have expanded the clinician’s ability to localize disease and tailor therapy in adult patients.2,7
Dietary intervention, consistent of high-protein, low-fat, medium-chain triglycerides (MCTs), which bypass the lymphatic system via portal absorption, is the cornerstone management, refractory or diffuse cases might benefit from pharmacologic agents (e.g: octreotide or sirolimus). 2
The absence of standardized therapeutic guidelines and heterogeneity in clinical response highlights the need for individualized treatment approaches, besides long term monitoring of complications such as immunodeficiency-related infections or lymphoproliferative transformation.2,5,6
We herein present a case of adult-onset PIL from Palestine, and we aim to highlight the strategic integration of clinical, endoscopic, and histopathological findings to achieve a diagnosis and to demonstrate the effectiveness of a tailored dietary and pharmacological regimen, contributing to the limited evidence base for managing this complex condition in adults.
Methods
Literature Review: To discover previously reported cases of Primary Intestinal Lymphangiectasia (PIL), we conducted a comprehensive literature review, particularly focused on adult onset. Using the keywords in combination with Boolean like primary intestinal lymphangiectasia, adult, protein-losing enteropathy, Waldmann’s disease, chylous ascites, and intestinal lymphangiectasia, as well as Palestine to know if there were any regional reports and so identify them, We searched the databases Scopus, PubMed, and Web of Science.
We found that no prior cases from Palestine were identified, emphasizing that this is the first ever reported case in the country. Highlighting how rare the disease is, the need for greater clinical awareness, and the importance of regional documenting to improve the global literature.
Case Presentation
A 28-year-old female presented with recurrent abdominal distension and chylous ascites. Her medical history was notable for a presumptive diagnosis of Crohn’s disease 7 years prior to presentation, based on clinical symptoms but without definitive endoscopic or histopathologic confirmation. Since 2021, she had experienced multiple episodes of ascites requiring therapeutic paracentesis.
7 years before presentation, the patient suffered nonspecific gastrointestinal symptoms, including diffuse recurrent intermittent crampy abdominal pain more prominent periumbilically and at the right lower quadrant, bloating, loose stools, and weight loss, raising suspicion for inflammatory bowel disease. Given the chronicity of symptoms for 3 years, Crohn’s disease was considered the most likely diagnosis at the time. However, during initial colonoscopy, no definitive lesions or histological features—such as granulomas, crypt distortion, or transmural inflammation—were identified. Despite the absence of endoscopic features related to Crohn’s disease, she was empirically managed as a case of Crohn’s for a short period with mesalamine and dietary modification, which provided limited symptomatic relief. Over time, the absence of progression, lack of classic Crohn’s complications (e.g., strictures, fistulas) and new-onset chylous ascites prompted reconsideration of the diagnosis. The working diagnosis of Crohn’s was ultimately abandoned in favor of further investigation into alternative causes of protein-losing enteropathy and ascites.
In early 2025, the patient presented with progressive abdominal fullness. She denied associated constitutional or gastrointestinal symptoms, including fever, night sweats, weight loss, diarrhea, or overt gastrointestinal bleeding. On physical examination, she was hemodynamically stable. Her abdomen was distended with shifting dullness, consistent with ascites. There was no evidence of peripheral edema, lymphadenopathy, or hepatosplenomegaly.
Laboratory investigations revealed a low serum albumin level of 2.3 g/dL, with normal serum immunoglobulin G (IgG) levels. Liver function tests were normal. Complete blood count was unremarkable. Serologic testing for Brucella and Widal was negative. A comprehensive tuberculosis workup, including interferon-gamma release assay (IGRA), was also negative. Stool analysis and cultures were negative for infectious pathogens. Ascitic fluid was chylous in appearance, sterile on culture, and negative for malignant cells on both cytology and cell block. A few reactive mesothelial cells were observed. Peripheral eosinophil count was not elevated, and autoimmune workup was unremarkable. A PET-CT scan was ordered for excluding occult malignancy, autoimmune, inflammatory or metabolically active conditions.
Abdominal CT imaging revealed diffuse thickening of the hepatic flexure of the colon, associated mesenteric fat stranding, and lymphadenopathy (largest node 1.5 cm). In addition to moderate-volume ascites, endoscopic evaluation (Figure 1) demonstrated multiple white plaques in the duodenum on upper endoscopy, while colonoscopy revealed no gross mucosal abnormalities. Biopsies were obtained from the duodenum, terminal ileum, colon, and stomach. Histopathologic examination of the duodenum revealed mild lymphatic dilation and mild eosinophilic infiltration (10 eosinophils/high-power field [HPF]) with preserved villous architecture. No parasites, granulomas, dysplasia, or malignancy were noted. Terminal ileal biopsies showed marked lymphatic dilation and a more intense eosinophilic infiltrate (25 eosinophils/HPF). Colonic biopsies demonstrated mild lamina propria edema and eosinophilia (20 eosinophils/HPF), without granulomas or neoplasia. Gastric mucosa appeared normal, with no Helicobacter pylori, intestinal metaplasia, or dysplasia. Upper gastrointestinal endoscopy showing multiple white plaques in the duodenum, suggestive of lymphatic dilation. These mucosal changes were most prominent in the second part of the duodenum and raised initial concern for infiltrative or lymphatic pathology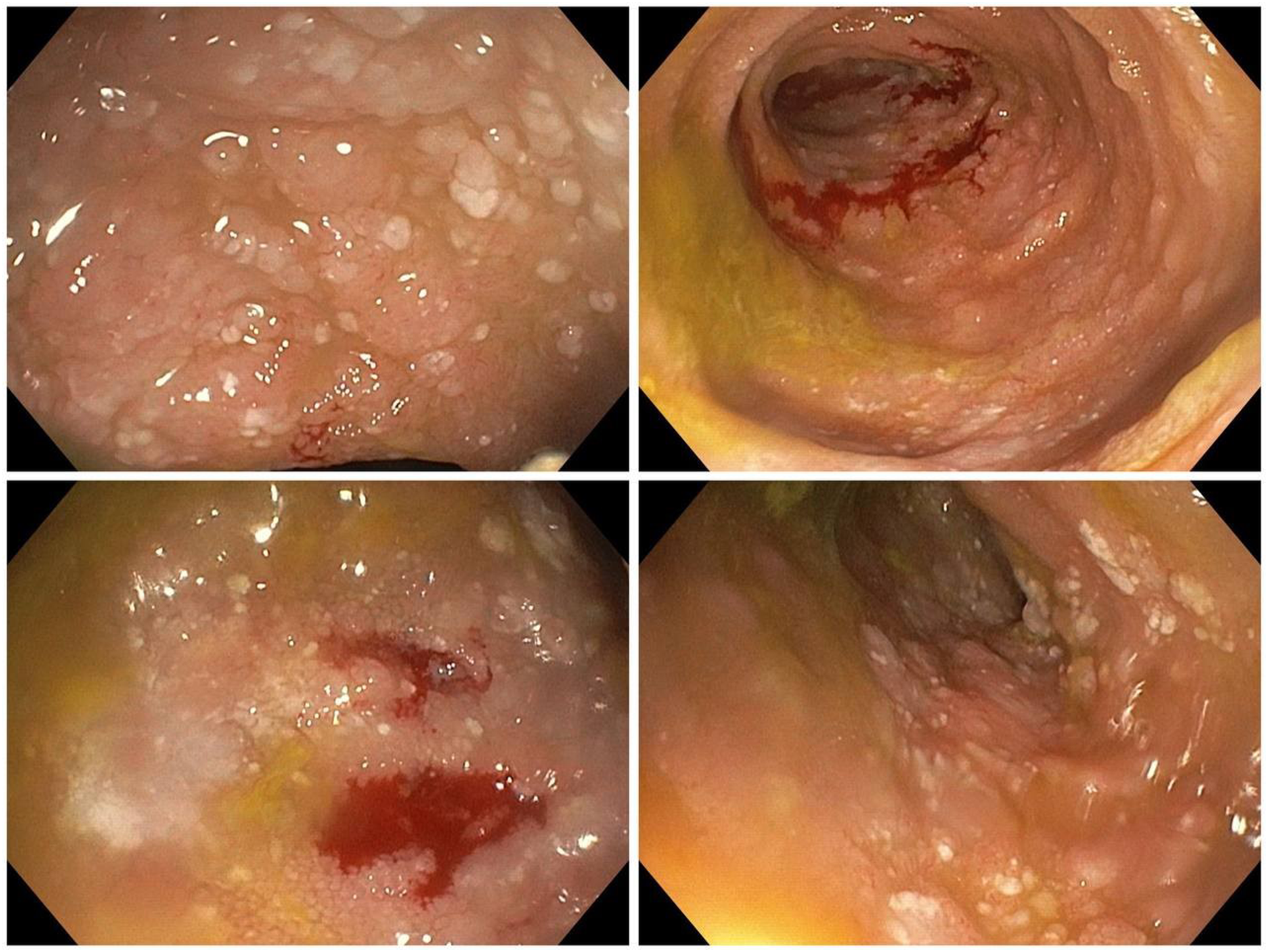
Final Diagnosis
The histological findings (Figure 2) were most consistent with Primary Intestinal Lymphangiectasia (PIL), characterized by dilated intestinal lymphatics, particularly in the ileum. The absence of granulomas, malignancy, parasites, or systemic infection helped exclude other differential diagnoses such as eosinophilic enteritis or secondary lymphangiectasia. (A)- Terminal ileum biopsy, the terminal ileal mucosa shows dilated lymphatic channels (H&E, 20x). (B)- The deep mucosal lymphatics are also dilated (H&E, 40x)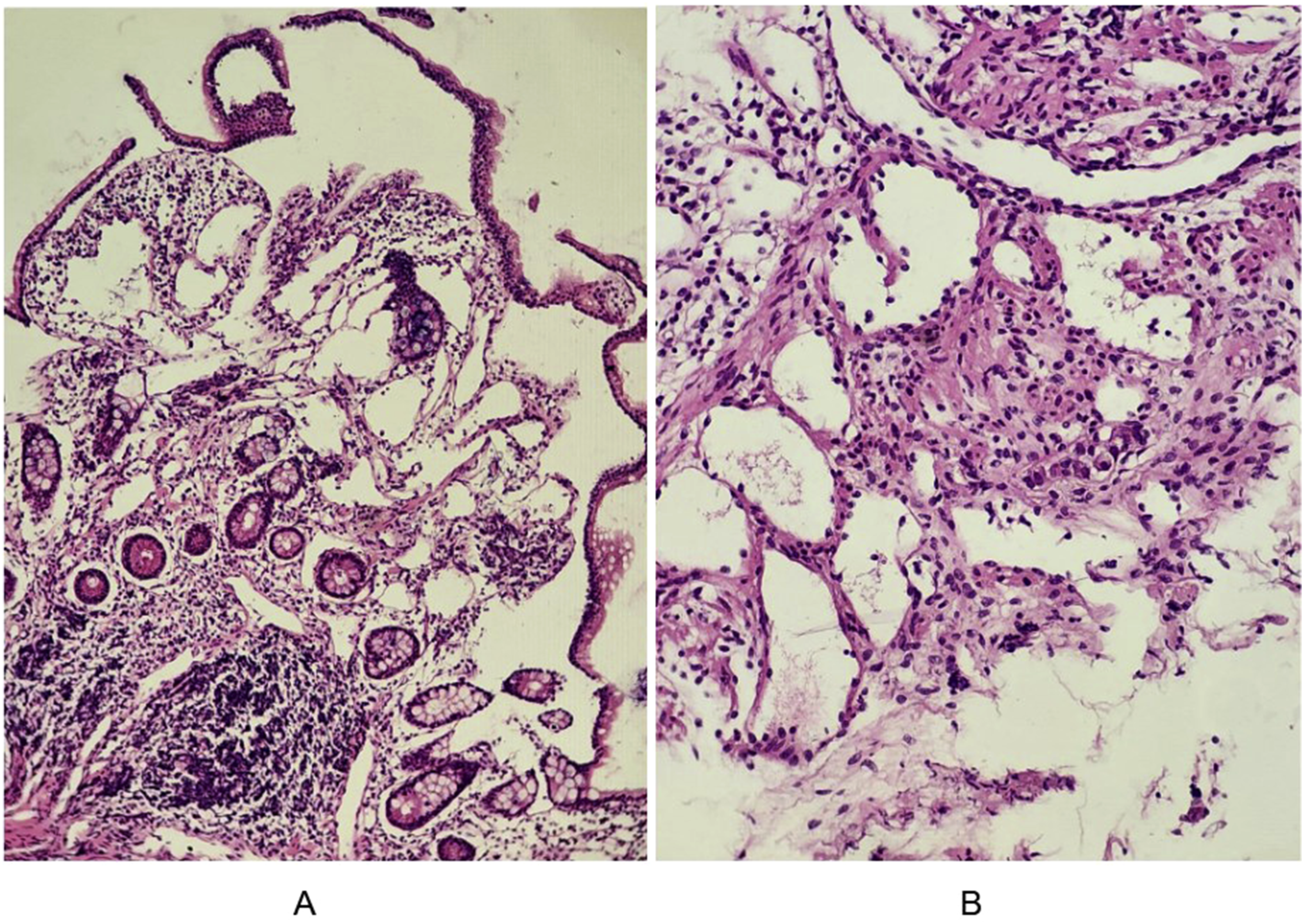
A whole-body PET/CT scan (Figure 3) performed a few days later demonstrated diffuse hypermetabolic bowel wall thickening involving the jejunum, ileum, ascending colon, hepatic flexure, and proximal transverse colon. This was accompanied by pericolonic fat stranding and mild-to-moderate fluorodeoxyglucose (FDG) uptake (Maximum Standardized Uptake Value - SUVmax = 5.5), without evidence of discrete lesions. A small hypodense liver lesion in segment VI (1.1 × 0.5 cm) exhibited FDG uptake. Non-metabolically active pelvic ascites was present. Several small mesenteric lymph nodes were noted (largest 1.3 × 0.7 cm) but without FDG avidity. No hypermetabolic lymphadenopathy was seen in the mediastinum, abdomen, or pelvis. FDG distribution in other organs was physiological. Whole-body PET/CT scan demonstrating diffuse FDG uptake in the jejunum, ileum, ascending colon, and proximal transverse colon, consistent with hypermetabolic bowel wall thickening. No discrete focal lesion or metabolically active lymphadenopathy was identified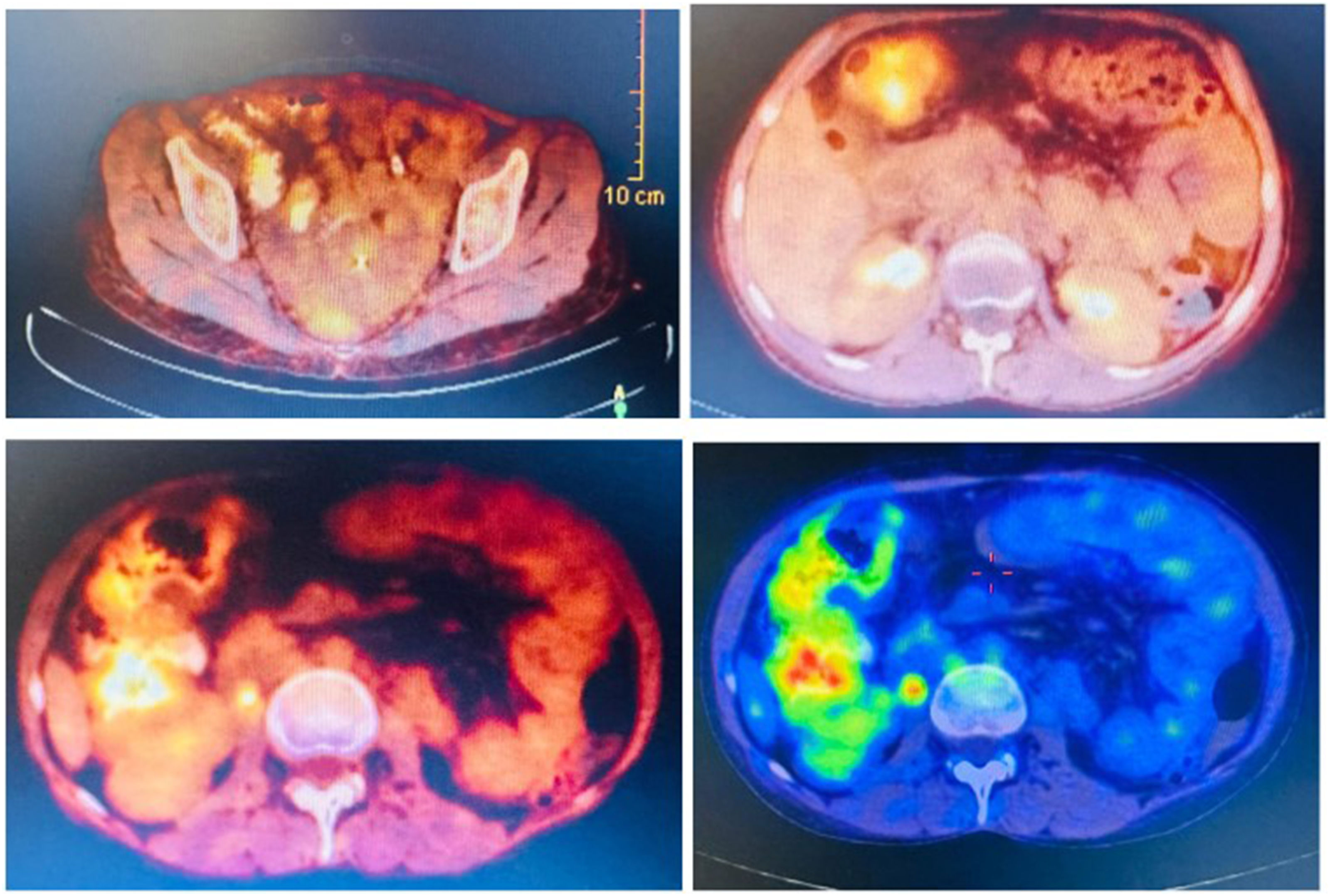
Diagnosis of PIL was made, and the patient was started on budesonide and long-acting octreotide (Sandostatin LAR 20 mg intramuscularly every 28 days). After 6 weeks, she showed clinical improvement with this therapy, with reduction in abdominal distension and a decreased need for paracentesis.
At the time of writing, the patient remained under outpatient follow-up. She had clinically improved, and further nutritional and immunologic assessment was planned, including dietary counseling and monitoring of protein-losing enteropathy. The combination of clinical, endoscopic, histologic, and imaging data supported the diagnosis of PIL with no evidence of malignancy or systemic inflammatory disease.
Discussion
Intestinal lymphangiectasia (IL) is a rare form of protein-losing enteropathy in which lymph spills into the intestinal lumen due to the rupture of the pathologically dilated intestinal lacteals (lymphatic vessels). Subsequent to the loss of protein-rich lymph, several laboratory abnormalities develop including hypoproteinemia, hypoalbuminemia, hypogammaglobulinemia, and lymphocytopenia. Additionally, the condition compromises nutritional status, often presenting with deficiencies in minerals such as iron and calcium, as well as lipids and fat-soluble vitamins. 8
Summary of Cases in Literature

The etiology of intestinal lymphangiectasia (IL) is classified into primary and secondary forms. 8 Our patient was diagnosed with PIL, a rare congenital disorder of the lymphatic system. Although PIL typically presents in childhood, adult-onset cases are uncommon and often pose a diagnostic challenge. In such cases, exclusion of secondary causes is essential, particularly because secondary IL is now considered more prevalent than previously thought. 8
Secondary IL may result from chronic inflammatory diseases (e.g., Crohn’s disease, celiac enteropathy), infections, cardiovascular disorders, malignancies, or iatrogenic causes such as surgery or radiation. 8 However, in our patient, no evidence of these contributing factors was identified, supporting the diagnosis of idiopathic adult-onset PIL.
The underlying pathophysiology of PIL involves abnormalities in lymphatic vessel development or function, leading to rupture of dilated lacteals and lymph leakage into the intestinal lumen. Molecular studies suggest dysregulated lymphangiogenesis, with increased expression of Vascular Endothelial Growth Factor (VEGF) receptor-3 and Lymphatic Vessel Endothelial Hyaluronan Receptor 1 (LYVE-1), alongside decreased VEGF-C and VEGF-D expression, contributing to this dysfunction. 15 These alterations have implications that include not only protein absorption but also broader physiological processes such as immune regulation and nutrient metabolism. 8
Clinical Spectrum of Intestinal Lymphangiectasia
The “classic” presentation of intestinal lymphangiectasia (IL) represents only the most severe end of a broad clinical spectrum observed in adults. 4 In its severe or classic form, IL may present with a variety of features, including edema—most notably lymphedema, which is less common, non-pitting, and identifiable by a positive Stemmer’s sign.4,8 This edema is often due to hypoalbuminemia. 8 Patients may also develop effusions characterized by the accumulation of chylous (lymph-filled) fluid in body cavities, such as the peritoneal space (chylous ascites), pleural cavity, and pericardial sac, gastrointestinal manifestations include chronic diarrhea due to fat malabsorption, often accompanied by steatorrhea, along with weight loss, muscle wasting, and general weakness.4,8
Other presentations may include abdominal masses, typically benign cystic lymphangiomas in the small or large bowel, as well as iron deficiency anemia, possibly related to chronic occult bleeding from associated small bowel angiodysplasia. 8 However, the mild or asymptomatic form of IL is more common in adults. With the advent of modern endoscopy, IL is now frequently identified incidentally in patients who display few or no clinical symptoms of malabsorption; supporting this, one cited study found histologically confirmed IL in 1.9% of 1,866 consecutive endoscopic examinations, with these patients showing no clinical signs of malabsorption even after one year. 4
Atypical Presentations and Complications
Our patient presented with chylous ascites, a striking but less frequent manifestation that was reported in some other cases.6,12,16 Several atypical presentations were also observed, expanding the phenotypic variability of PIL: one patient had isolated seizures due to hypocalcemia, without any gastrointestinal symptoms 14 ; another presented with widespread HPV-related warts due to significant cellular immunodeficiency 17 ; and a unique case showed complete symptom remission during pregnancy and oral contraceptive use, suggesting a possible hormonal influence 6 Other rare findings included intestinal obstruction due to solitary lymphangiectasia, 18 colonic microperforation, 19 tuberculosis infection as a complication, 20 and lymphoma transformation after 23 years of chronic disease. 21
Diagnostic Markers and Challenges
Hypoalbuminemia is a consistent diagnostic marker across IL cases (Table 1), reflecting lymphatic leakage. 8 Lymphopenia and hypogammaglobulinemia (particularly low IgG) are frequent,9,17,20,21 though our patient had normal lymphocyte counts and IgG, aiding exclusion of hepatic or immunodeficiency-related causes. Vitamin deficiencies and neuromuscular complications, such as seizures, are also reported.10,14 Cellular immune dysfunction increases risks of opportunistic infections (e.g., HPV warts, tuberculosis), warranting caution with live vaccines.17,20 The diagnostic challenge lies in IL’s variable presentation and overlap with other conditions, as seen in our patient’s initial Crohn’s misdiagnosis. Excluding secondary causes and confirming histological findings (e.g., dilated lymphatics) are critical for accurate diagnosis. 8
The initial suspicion of IL should arise in the presence of lymphocytopenia, hypogammaglobulinemia, and hypoalbuminemia. Definitive diagnosis, however, requires both characteristic endoscopic and histopathological features. Endoscopically, IL may present with edematous, whitish villi that give a “snowflake appearance,” often accompanied by scattered white plaques. 22 Histologically, the hallmark finding is dilated lacteals within the lamina propria of the small intestinal mucosa, in the absence of villous atrophy or infectious etiologies.4,22
In our patient, endoscopy revealed typical plaques in both the duodenum and terminal ileum.15,23 Histopathology confirmed mild lymphatic dilation in the duodenum and marked lymphatic dilation in the terminal ileum, findings consistent with the segmental and often patchy nature of IL. 22 This aligns with reports that IL may predominantly affect the ileum and that superficial duodenal biopsies may underestimate disease severity due to deeper or segmental involvement. 4 The presence of tissue eosinophilia across multiple sites (duodenum, ileum, colon) is likely a reactive phenomenon secondary to lymph extravasation, rather than indicative of a primary eosinophilic gastrointestinal disorder—particularly in the absence of villous atrophy, crypt hyperplasia, or granulomatous inflammation.
Moreover, our diagnostic approach is consistent with the framework suggested by Kwon and Kim, which emphasizes confirming protein-losing enteropathy, excluding secondary causes such as cardiac disease or malignancy, and using imaging to evaluate disease extent. 22 Although, for our patient, advanced imaging modalities like MR lymphangiography were not available, CT scanning and histology provided sufficient diagnostic support. Along with the relevant laboratory abnormalities, endoscopic findings, and histopathological confirmation—particularly the significant lymphatic dilation in the ileum— this combination established the diagnosis of intestinal lymphangiectasia in this patient, despite its variable and focal nature.
In literature, diagnosis was frequently delayed due to the subtle nature of symptoms and the limited yield of conventional endoscopy.9,11,16 Advanced imaging and endoscopic modalities, including capsule endoscopy, double-balloon enteroscopy, and push enteroscopy, facilitated better visualization of white villi, mucosal edema, and dilated lacteals.6,9,11-13 Histological confirmation using D2-40 immunostaining consistently demonstrated dilated mucosal and submucosal lymphatics, making it the gold standard diagnostic method (Table 1). Additionally, CT and MR imaging revealed bowel wall thickening and lymphatic abnormalities in several patients,6,10,12,13,16 while lymphoscintigraphy or MR lymphangiography helped confirm lymphatic leaks in diagnostically challenging cases.6,9
The cornerstone of treatment for primary intestinal lymphangiectasia (PIL) is dietary modification, specifically a high-protein, low-fat diet enriched with medium-chain triglycerides (MCTs), which are absorbed directly into the portal circulation, bypassing the diseased lymphatics and thereby reducing lymphatic pressure.4,22 This approach was effective in the majority of reported cases, including ours, with most patients demonstrating clinical improvement on dietary therapy alone (Table 1). In cases refractory to dietary management, further treatment depends on the extent of disease involvement. 22 Focal disease may respond well to localized interventions such as surgical resection or radiologic lymphatic embolization,18,19,22 while diffuse intestinal involvement typically necessitates systemic therapy. Medical treatments such as octreotide—a somatostatin analogue—have shown benefit,9,13,22 and other agents like sirolimus and corticosteroids have been used successfully in selected cases. 9 Additionally, patients with secondary causes of intestinal lymphangiectasia, such as portal hypertension due to cirrhosis, may be managed conservatively. 16
Several complications were noted across cases. Thromboembolic events, such as pulmonary embolism and cerebral infarction, occurred in the setting of severe hypoalbuminemia and protein loss, such as antithrombin III alongside albumin loss. 9 Immunodeficiency-related infections were common, including cellulitis, 9 generalized HPV, 17 and tuberculosis. 20
The most severe reported complication was intestinal lymphoma, diagnosed in a patient after 23 years of PIL. 21 These findings underscore the importance of long-term monitoring, including nutritional assessment, immunologic evaluation, and malignancy screening. Our patient has shown early response to dietary therapy, with no complications reported thus far.
In our patient, the diagnostic journey underscored several critical knowledge gaps that continue to challenge clinicians managing intestinal lymphangiectasia. Despite presenting clinical and biochemical features suggestive of the disease, the absence of standardized approaches to quantify lymphatic dysfunction made it difficult to objectively assess disease burden or monitor response to therapy.
The lack of early biomarkers further delayed definitive diagnosis, emphasizing the need for predictive tools that can identify patients at risk before irreversible complications develop.
This case also reflects broader gaps in our understanding of the disease’s natural history, particularly in adult-onset presentations, which remain poorly characterized. The clinical heterogeneity observed across patients suggests that individualized diagnostic and therapeutic strategies are urgently needed. Continued research is essential to uncover the underlying mechanisms driving variability in presentation and response to treatment, and to ultimately guide the development of more targeted, personalized care.
Conclusion
PIL is a rare condition in adults that can mimic more common gastrointestinal disorders, leading to delayed diagnosis. This case highlights the importance of considering PIL in patients with unexplained chylous ascites and hypoalbuminemia. Diagnosis relies on histopathologic confirmation and exclusion of secondary causes. Dietary management is the cornerstone of treatment, with adjunctive therapies reserved for refractory cases.
Reporting this adult-onset case adds to the limited literature and emphasizes the need for early recognition to improve patient outcomes; this is the first case in Palestine to our knowledge, and that there is a need for better documentation of such cases, to assess the prevalence of the disease and patients characteristics in Palestine comparing with other demographics.
Footnotes
Acknowledgement
The authors would like to extend their thanks to the patient for agreeing to share her case and wish her a full recovery.
Ethical Considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Consent for Publication
Verbal informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.