
Abstract
Systemic lupus erythematosus (SLE) is an autoimmune connective tissue disorder characterized by a wide spectrum of clinical features ranging from skin and joint involvement to organ dysfunction, including the central nervous system, peripheral nervous system (PNS), and renal failure. Guillain–Barré syndrome (GBS) is an immune-mediated acute polyradiculoneuropathy involving the PNS and represents an unusual initial manifestation of SLE. An 18-year-old female presented with subacute, progressive, ascending weakness of the lower and upper limbs, accompanied by hyporeflexia. Further clinical and laboratory evaluation revealed that the patient fulfilled the diagnostic criteria for SLE. Electromyography and nerve conduction studies demonstrated normal conduction velocities and latencies, but there was a reduced amplitude of compound motor action potentials in the distal muscles of the upper and lower limbs, without conduction block. Needle electromyography findings were consistent with a diagnosis of acute motor axonal neuropathy, considered a moderate-to-severe form of GBS. On evaluation, the patient exhibited polyarthralgia, widespread generalized erythematous rash, microcytic anemia, and bilateral pleural effusion. Immunological profiling revealed high-titer antinuclear antibody (1:1280), elevated anti-dsDNA (171 IU/mL), positive anti-Sm antibodies, and low C3/C4 levels. Treatment with pulse steroids (methylprednisolone 1 g/day for 3 days), immunosuppressants, and intravenous immunoglobulin led to marked improvement. This case emphasizes that SLE can precipitate GBS, and early recognition of this overlap may influence therapeutic strategies and improve outcomes.
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune connective tissue disorder that can involve a wide range of organs, including the skin, joints, kidneys, lungs, heart, and peripheral nervous system (PNS). It is influenced by genetic, immunological, and environmental factors. 1 Central nervous system disease is one of the most frequent complications and may occur at any stage of SLE, whereas PNS manifestations are infrequent, typically presenting as distal symmetric axonal polyneuropathy or multiple mononeuropathies. 2
Guillain–Barré syndrome (GBS) is a rare autoimmune polyradiculoneuropathy in which the immune system targets peripheral nerves, producing symptoms such as paresthesia, weakness, and in severe cases, paralysis with respiratory involvement. Although GBS has been described in association with SLE, its occurrence as an initial manifestation is uncommon. It accounts for fewer than 10% of neuropsychiatric SLE presentations, and its incidence among patients with SLE has been reported to be ~1.5%.3,4 Here, we present a patient with GBS as the first clinical manifestation of SLE.
Case Presentation
An 18-year-old female presented with new-onset, progressive motor weakness involving all 4 extremities in an ascending pattern. Over the 10 days preceding admission, she experienced increasing difficulty in standing and walking. There were no sensory symptoms at the onset. Two weeks prior to admission, she had developed a flu-like illness with fever, night sweats, and purulent discharge from both ears. Around the same time, she also noticed a generalized skin rash (Figure 1A and B). Upon admission, her weakness worsened further. She began to experience paresthesia and progressive weakness beginning in the hands, spreading to involve the upper limbs and then the lower limbs, significantly impairing her ability to perform daily activities. The weakness was not associated with other focal neurological deficits, and there was no evidence of respiratory muscle involvement. She denied any history of recent gastroenteritis, travel, or immunizations.

(A) Widespread erythematous maculopapular rash with fine scaling over the abdomen and lower torso, consistent with cutaneous involvement suggestive of lupus dermatitis. (B) Multiple hypopigmented macules over the forehead, some coalescing (arrow), these lesions may indicate discoid lupus.
Systemic review revealed a dry cough and pleuritic chest pain, but no other systemic symptoms. Her drug, social, and menstrual histories were unremarkable. On general examination, she appeared pale. Her vital signs were stable: SpO2 was 98%, pulse rate 80 beats/minute, and blood pressure 110/70 mmHg. Neurological examination: She was alert and oriented with intact mental status. Cranial nerves II to XII were grossly intact. Upper limbs showed no muscle wasting or fasciculations, but hypotonia was present. Muscle strength was reduced to Grade 2 proximally and Grade 3+ distally. In the lower limbs, tone was normal with no muscle wasting. Power was Grade 3+ proximally and Grade 4 distally. Deep tendon reflexes were diminished (1+) in the upper and lower limbs, and the sensation was preserved throughout both upper and lower limbs. Musculoskeletal examination showed bilateral diffuse knee swelling with mild warmth, a positive ballottement sign, and mild restriction in all ranges of motion. Cardiovascular examination was unremarkable with normal heart sounds and no added sounds, thrills, or heaves. Respiratory examination revealed normal chest shape and expansion, but bilateral dullness to percussion at the lung bases, harsh vesicular breath sounds, and reduced air entry bilaterally.
Laboratory investigations showed a hemoglobin level of 8.2 g/L (normal: 12-16 g/dL), white blood cell count of 4.29 × 103/μL (normal: 4.5-11 × 103/μL), and platelet count of 294 × 103/μL (normal: 150-450 × 103/μL). C-reactive protein was elevated at 51 mg/L (normal: <5 mg/L). Renal function tests were normal with a serum creatinine of 0.44 mg/dL (normal: 0.6-1.2 mg/dL) and urea of 29 mg/dL (normal: 7-20 mg/dL). Iron profile showed serum iron at 6.26 mg/dL (normal: 50-170 μg/dL) and ferritin at 58.83 mg/dL (normal: 12-150 ng/mL in females). Lactate dehydrogenase was 259 U/L (normal: 140-280 U/L) and serum vitamin B12 was 412 pg/mL (normal range: 200-900 pg/mL). Liver function tests (aspartate aminotransferase: 10-40 U/L, ALT: 7-56 U/L, ALP: alkaline phosphatase: 44-147 U/L, total bilirubin: 0.3-1.2 mg/dL, albumin: 3.5-5.0 g/dL), thyroid function (TSH: 0.4-4.0 mIU/L, free T4: 0.8-1.8 ng/dL), procalcitonin (<0.05 ng/mL), serum electrolytes (Na⁺: 135-145 mmol/L, K⁺: 3.5-5.0 mmol/L, Cl−: 98-107 mmol/L, HCO3−: 22-29 mmol/L, Ca2+: 8.5-10.5 mg/dL), prothrombin time (11-13.5 s), partial thromboplastin time (25-35 s), and international normalized ratio (0.8-1.2) were all within normal limits. Blood cultures showed no growth. Immunological tests revealed a positive antinuclear antibody (ANA; 1:1280), high anti-dsDNA level (171 U/mL, normal: <30 U/mL), strong positive anti-Sm, positive PCNA, and low C4 levels (normal: 10-40 mg/dL). Serum cryoglobulin was absent (Table 1). Infectious serologies for hepatitis B, hepatitis C, cytomegalovirus, and HIV were all negative. Imaging studies included a cervical MRI which was normal except for circumferential disc bulging at C5 to C6 (Figure 2). A chest computed tomography scan showed bilateral pleural effusions, bilateral axillary lymphadenopathy, and bilateral ground-glass opacities with normal vascular markings. Abdominal ultrasound revealed hepatomegaly measuring 18 cm and bilateral pleural effusion. Electromyography and nerve conduction studies performed on day 6 of admission revealed normal conduction velocities and latencies, but there was a reduced amplitude of compound muscle action potentials in the distal upper and lower limb muscles, without evidence of conduction block. Also she underwent a lumbar puncture, which revealed albuminocytologic dissociation (protein 134 mg/dL, normal: 15-45 mg/dL, WBC 3 cells/μL, normal: <5 cells/μL). Needle electromyography findings supported the clinical diagnosis of acute motor axonal neuropathy, a moderate-to-severe variant of GBS. Repetitive nerve stimulation did not show significant decrement in motor responses. There was no electrophysiological evidence of neuromuscular junction disorders or primary myopathies.
Basic Laboratory Investigation.
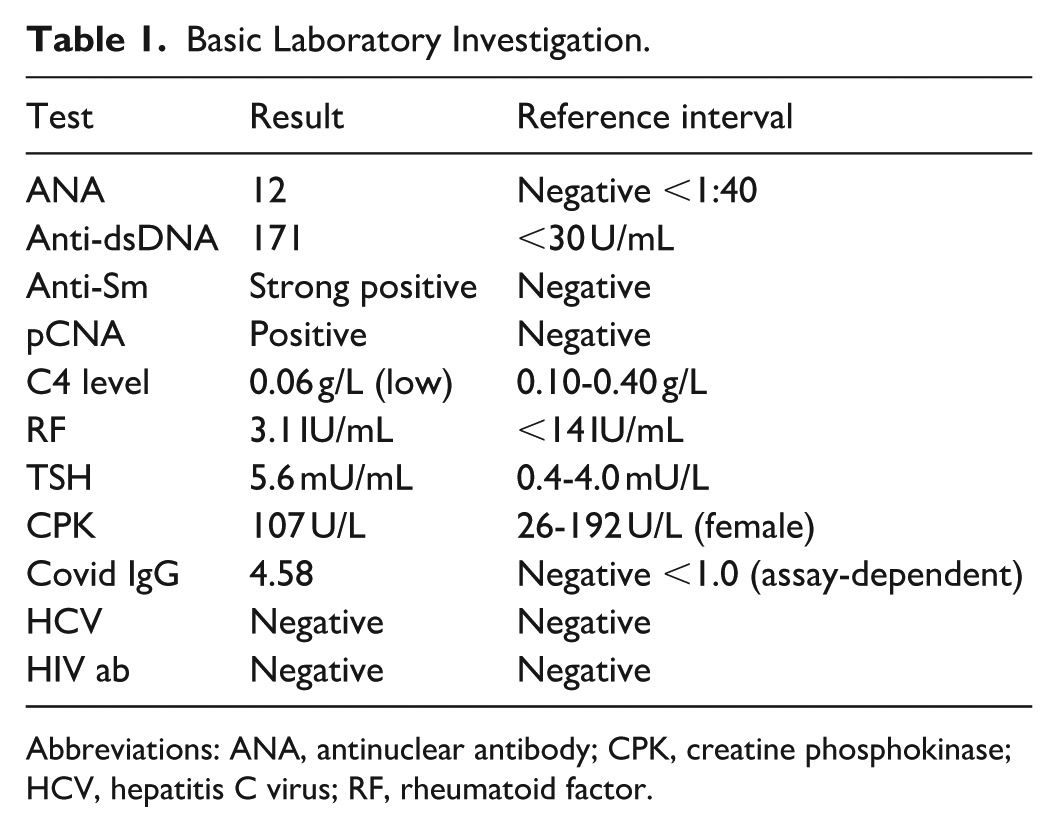
Abbreviations: ANA, antinuclear antibody; CPK, creatine phosphokinase; HCV, hepatitis C virus; RF, rheumatoid factor.

MRI of the cervical spine showing mild degenerative changes (arrow), including loss of lordosis and a C5 to C6-disc bulge, with normal vertebral alignment and spinal cord.
Initially, the patient underwent 4 sessions of plasma exchange (PLEX), but there was no significant clinical improvement. She was then treated with pulse steroids (methylprednisolone 500 mg IV daily for 5 days), intravenous immunoglobulin (IVIG) at 2 g/kg over 3 days, and immunosuppressive therapy. Follow-up 4 days later revealed significant improvement in muscle strength, now Graded 4 in both upper and lower limbs, and the reflexes were preserved.
Discussion
SLE is a multisystem autoimmune disorder characterized by diverse clinical features that may include renal, hematologic, and neuropsychiatric involvement. Although PNS manifestations in SLE are uncommon, they are being increasingly reported, with GBS representing one of the rare but clinically important associations.3,5 The occurrence of GBS as the first clinical manifestation of SLE is exceptionally rare and highlights the need for prompt recognition and precise diagnosis. Classically, GBS is an acute immune-mediated neuropathy involving the PNS, most often triggered by preceding infection, vaccination, or, on rare occasions, another autoimmune condition such as SLE.6-8
In our case, a previously healthy 18-year-old female presented with new-onset, progressive motor weakness in all 4 extremities, initially without sensory involvement along with hyporeflexia and electromyography findings all supporting and consistent with GBS. The temporal relationship between her flu-like illness and the subsequent neurological symptoms raised suspicion for an autoimmune etiology. A development of joint symptoms, skin rash, lung involvement, and laboratory workup revealed positive ANA, anti-dsDNA, anti-Sm antibodies, and low C4 levels, supporting the diagnosis of SLE. This is consistent with previous case reports where GBS was found to be the initial manifestation of SLE.7,9 The pathophysiology behind the association between GBS and SLE remains unclear, but it is hypothesized that the autoimmune antibodies in SLE may predispose patients to GBS through target myelin tissue, contributing to peripheral nerve damage which could lead to the development of both diseases concurrently.10,11
Although there are well-established guidelines for the diagnosis and management of GBS (Leonhard et al), but no specific guidelines for GBS occurring in the context of SLE.12,13 Treatment modalities for GBS occurring in the setting of SLE have included corticosteroids, cyclophosphamide, plasmapheresis, immunoglobulin therapy, and other immunosuppressive agents. 14 Gao et al suggested using intravenous pulses of methylprednisolone for 5 days in combination with monthly pulses of cyclophosphamide. 5 Our patient’s treatment course was complicated by a lack of significant improvement after PLEX therapy, which is typically used for GBS treatment. After switching to pulse steroids and IVIG, followed by immunosuppressive therapy, the patient showed significant improvement. This therapeutic approach aligns with findings in other case reports where a combination of immunotherapy is required to manage both GBS and the underlying SLE.5,15 Interestingly, a comparable case in a pediatric patient demonstrated that IVIG and corticosteroids were effective in SLE-associated GBS, although additional treatment with rituximab and prolonged ventilation was also required. 9 While GBS occurring as the initial manifestation of SLE is very uncommon, such an association should be considered, particularly in young patients who present with acute neurological deficits. A strong clinical suspicion and prompt management are essential to prevent irreversible damage and improve prognosis.8,16 In our patient, early recognition followed by treatment with steroids, IVIG, and subsequent immunosuppressive therapy was crucial for recovery, highlighting the importance of individualized therapeutic strategies in complex SLE-GBS cases.
Conclusion
GBS may rarely occur as an initial presentation of SLE, and this poses a diagnostic challenge. It should therefore be considered when patients, especially young females, present with neurological features suggestive of autoimmune neuropathy. Clinicians are advised to keep a strong clinical suspicion for underlying SLE in unusual or refractory cases of GBS. Timely recognition and initiation of therapy, including IVIG, corticosteroids, and immunosuppressants, are crucial for improving patient outcomes. Moreover, additional research is warranted to clarify the underlying mechanisms connecting GBS and SLE and to establish standardized management strategies for such rare presentations.
Footnotes
Acknowledgements
The authors would like to thank the Rheumatology, Neurology, and Radiology teams at the Medical City Baghdad Teaching Hospital for their efforts in managing the patient. We are also grateful to the staff of the Emergency, Laboratory, and Radiology departments for their valuable assistance in the investigations. Finally, we extend our special thanks to the patient and her family for their trust, cooperation in the care provided, and their kind permission to publish this case report for educational purposes.
Ethical Considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Consent for Publication
Written informed consent was obtained from the patient family for their anonymized information to be published in this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.