
Abstract
A 46-year-old man with de novo acute myeloid leukemia (AML) M1 achieved complete remission after induction therapy and received consolidation treatment. Due to intermediate-risk classification and positive minimal residual disease at 6 months after consolidation treatment, he underwent haploidentical hematopoietic stem cell transplantation (HSCT) from his son. Post-transplant, he had successful neutrophil and platelet recovery without transfusion support and experienced cytomegalovirus reactivation that was effectively managed. On day +108 post-transplant, he developed fever and diarrhea. The differential diagnosis included opportunistic infection, neoplastic infiltration, and chronic on acute gastrointestinal (GI) graft-versus-host disease, given the timeframe and clinical presentation. Further investigations, including imaging and biopsies, led to a diagnosis of extranodal NK/T-cell lymphoma (ENKL). Despite adjustments in therapy, his condition worsened, and he succumbed to GI bleeding and multiorgan failure on day +144 post-transplant. This case illustrates the rare occurrence of ENKL following haploidentical HSCT in an AML patient, highlighting the diagnostic challenges and the highly aggressive clinical course of ENKL. It reinforces the importance of vigilant post-transplant monitoring, not only for infectious and relapse-related complications but also for secondary lymphoid malignancies and unusual presentations, to enable prompt diagnosis and timely intervention.
Keywords
Introduction
Post-transplant lymphoproliferative disorder (PTLD) manifests as a proliferation of lymphoid or plasmacytic cells. This condition arises as a result of immunosuppression in patients who have undergone allogeneic hematopoietic stem cell transplantation (HSCT) or solid organ transplantation. PTLD is considered among the most grave complications post-transplantation, attributed mainly to its high mortality rate.1,2 Multiple studies have shed light on the epidemiology and risk factors associated with the development of PTLD following allogeneic HSCT. It commonly develops within the first year post-transplantation, with an overall incidence rate of approximately 3%. 3 Several prominent risk factors have been identified for PTLD after allogeneic HSCT. These include the employment of a reduced intensity conditioning regimen, the type of donor (such as an unrelated fully matched or partially mismatched donor or a sibling mismatched donor), mismatches in Epstein–Barr virus (EBV) serology between donor and recipient (notably, a negative recipient and a positive donor), prophylactic strategies for graft versus host disease (GVHD), which involves in vivo or ex vivo T cell depletion, the use of anti-thymocyte globulin (ATG) or alemtuzumab, and the utilization of peripheral blood stem cells as the stem cell source. 4
Herein, we report a rare case of EBV-related extranodal NK/T cell lymphoma (ENKL) presenting with hepatic lesions 4 months after transplantation.
Case Presentation
A 46-year-old man was diagnosed with de novo acute myeloid leukemia (AML) M1 in 2021 according to the French-American-British classification. 5 Cytogenetic examination at diagnosis revealed a normal karyotyep. Additional molecular testing revealed no mutations in fms-related tyrosine kinase 3 internal tandem duplication, as well as in nucleophosmin 1 and CCAAT/enhancer-binding protein alpha (CEBPA). After initial induction therapy with the 7+3 regimen, he achieved complete remission (CR) and then received high-dose cytarabine as consolidation treatment. Since the patient lacked a fully matched donor for HSCT and was classified as having intermediate risk based on the European Leukemia Network 2022 genetic risk stratification, 6 he underwent follow-up every 3 months to assess minimal residual disease (MRD) through bone marrow (BM) flow cytometry, in accordance with our center’s practice and current expert recommendations,6,7 which support deferring allogeneic HSCT in CR1 for intermediate-risk AML patients in the absence of positive MRD. Six months later, flow cytometry detected a positive MRD, qualifying him for an allogeneic HSCT from any suitable donor at the earliest opportunity. In June 2023, the patient received a haploidentical HSCT from his 11-year-old son, 15 months following his AML diagnosis, at the Research Institute for Oncology, Hematology and Cell Therapy, Shariati Hospital. The conditioning regimen was composed of intravenous (IV) busulfan at 3.2 mg/kg/day administered from day −7 to −4, fludarabine at 30 mg/m2/day from day −7 to −3, and ATG at 2.5 mg/kg/day from day −3 to day −2. To prevent GVHD, cyclophosphamide was given intravenously at 40 mg/kg/day from day +3 to +4, and cyclosporine A at 3 mg/kg/day from day +5 to +15. During transplantation, the patient received 6.35 × 108/kg mononuclear cells, 8.54 × 106/kg CD34+ cells, and 243 × 106/kg CD3+ cells. Recovery of neutrophils (count >0.5 × 109/L) and platelets (count >20 × 109/L) occurred on days +14 and +17, respectively. On day +10, reactivation of cytomegalovirus (CMV) was identified despite prophylaxis with valacyclovir 1000 mg/day; the patient was administered valganciclovir at a dosage of 1800 mg/day for 5 days, then 900 mg/day for the next 35 days. On days +16 and +23, due to his MRD-positive status pre-HSCT and high risk of relapse, the patient was given infusions of allogeneic natural killer (NK) cells at dosages of 2 × 106 and 5 × 106 cells/kg, respectively, as part of a clinical trial assessing the infusion of allogeneic NK cells after HSCT in high-risk AML patients. During his recovery period at our facility, the patient developed stage I acute skin GVHD, which was managed with CSA. The patient was discharged on day +26. Initial BM aspiration and biopsy after the transplant, performed on day +50, revealed hypocellular marrow, a short tandem repeat (STR) analysis indicating 99% donor chimerism, and a negative MRD result from flow cytometry. In September 2023 (day +108), the patient presented to the emergency department (ED) with a chief complaint of fever and diarrhea (which was not green) from 20 and 10 days prior, respectively. Preliminary assessments in the ED comprised a spiral chest computed tomography (CT) scan and an abdominopelvic ultrasound examination. Spiral chest CT revealed multiple scattered nodular opacities with ground-glass halos in the upper lobes of both lungs (Figure 1A). Abdominopelvic ultrasound indicated several roundish hypoechoic lesions in the liver, the largest measuring 24 × 32 mm, without any abdominal lymphadenopathy. He was admitted to our post-BMT ward due to initial suspicions of post-transplant opportunistic infections or potential neoplastic infiltrations for additional evaluation, as there were no signs of skin GVHD and his tests for liver and renal function were normal. A subsequent triphasic abdominopelvic spiral CT scan confirmed multiple ill-defined hypodense lesions of varying sizes throughout the liver parenchyma, the largest measuring 20 mm, accompanied by mild ascites and splenomegaly (Figure 1B). Further evaluations showed that CMV was not detectable through real-time DNA polymerase chain reaction (qPCR), and the galactomannan test was negative. The patient was started on broad-spectrum antibiotics and antifungal medications. Although liver function tests were normal, given the suspicion of chronic on acute gastrointestinal (GI) GVHD, IV CSA and methylprednisolone were incorporated into the treatment plan. The patient’s fever and diarrhea subsided within a week after starting treatment, although there was an increase in the volume of his abdominal ascitic fluid during this time. Subsequently, the patient received an ultrasound-guided biopsy of the liver lesions following the placement of a nontunneled peritoneal drainage catheter to address his ascitic fluid, which was found to be hemorrhagic with a low serum-ascites albumin gradient upon analysis. On day +117, BM aspiration and biopsy demonstrated mildly hypocellular marrow with less than 3% blast cells. STR analysis showed 100% donor chimerism, and flow cytometry from the BM aspiration indicated negative MRD. The patient also had a bronchoalveolar lavage performed because of diffuse centrilobular nodules identified in the upper segments on the spiral chest CT scan. The smear showed no remarkable findings, but the culture revealed a polymicrobial presence. Additionally, EBV real-time quantitative PCR from the patient indicated 2704 copies/mL. Sputum smears and qPCR for acid-fast bacilli in ascites were negative, although the ascitic culture yielded polymicrobial results. Immunohistochemical analysis of the hepatic biopsy revealed that the atypical lymphoid cells expressed CD3, CD7, and CD56 but lacked expression of CD34, MPO, CD20, CD4, and CD8 (Figure 2). Furthermore, chromogenic in situ hybridization for Epstein–Barr virus-encoded small nuclear RNAs (EBER) was conducted, exhibiting a positive reaction in a subset of atypical lymphoid cells (Figure 3). These findings were consistent with a diagnosis of peripheral T-cell lymphoma, most likely ENKL. Moreover, ascitic flow cytometry detected NK/T-cells. Upon diagnosing ENKL, CSA was discontinued. At the same time, the methylprednisolone dosage was reduced to 20 mg per day. The patient was evaluated for the feasibility of treatment with the CHOP chemotherapy protocol. However, following the administration of 2 mg vincristine, the patient’s clinical condition deteriorated, leading to the cessation of chemotherapy. The build-up of ascitic fluid led to pleural effusion, causing respiratory distress. With the patient’s consciousness worsening, paraclinical examinations indicated a progression to a state of disseminated intravascular coagulation. Ultimately, in November 2023 (day +144), the patient succumbed to GI bleeding and ensuing multiorgan failure.

Imaging studies. (A) Multiple centriacinar nodular opacities at both lungs in the spiral chest CT scan. (B) Several variable-sized round-shaped lesions throughout the liver parenchyma on spiral CT scan of the abdomen were suggestive of an opportunistic infection or neoplastic infiltrations.
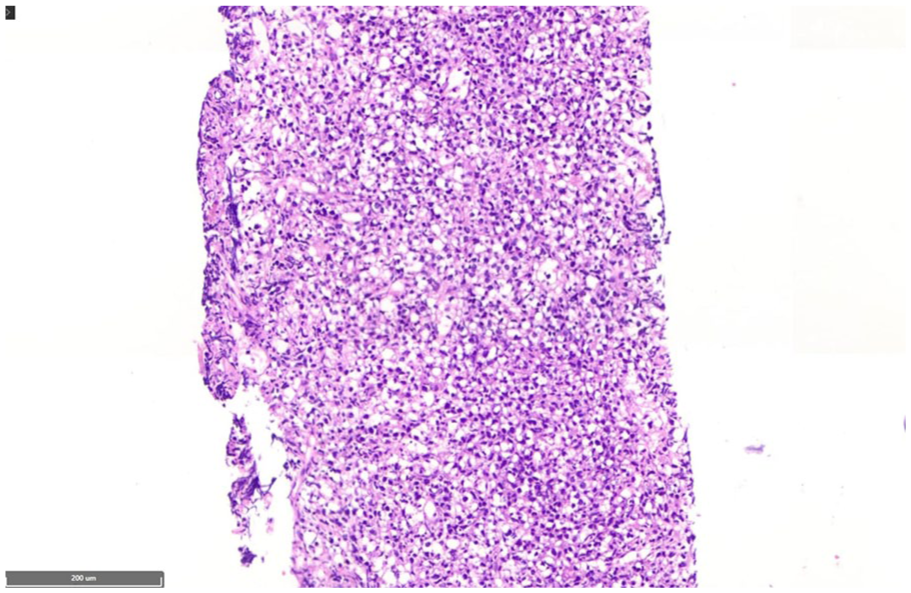
Histologic images of the biopsied liver revealed sheets of monomorphic medium-sized atypical lymphoid cells (hematoxylin and eosin staining, ×100).

Immunohistochemistry staining showed that the atypical lymphoid cells were positive for CD3, CD7, and CD56 (as shown here), and negative for CD20, CD4, and CD8 (×100). The overall findings are compatible with peripheral T-cell lymphoma, most likely NK/T-cell lymphoma.

EBER in situ hybridization showing nuclear staining in some atypical lymphoid cells ( × 200).
Discussion
PTLD comprises a diverse group of morphologically distinct entities that exhibit varying clinical manifestations. 8 It is characterized by prolonged fever, lymphadenopathy, periodic tonsil enlargement, and a decline in cellular lineages, as evidenced by complete blood count. 9 In our case, the patient presented with hepatic lesions as extranodular manifestations. While B-lineage PTLDs are the most prevalent, T- and NK-cell PTLDs (T/NK-PTLDs) are relatively uncommon, accounting for approximately 2–15% of all PTLD cases. 10 T/NK-PTLDs typically manifest at a later stage following transplantation and are associated with an unfavorable prognosis. Unlike their B-cell counterparts, only a subset of T/NK-PTLDs (up to 37%) are associated with EBV infection.10, 11 The median overall survival for T/NK-cell PTLDs is reported to be as short as 6 months. 10 Additionally, a T/NK-cell origin in PTLDs is recognized as a significant predictor of poor prognosis. 12
Two main factors contribute to the development of PTLD: (1) the EBV serological status of both the transplant recipient and donor and (2) the extent of T-cell immunosuppression before and after the transplant. Among these, EBV serological status is regarded as the most significant risk factor for PTLD following solid organ transplantation (SOT). 13 A mismatch in EBV serology between the recipient (R−) and donor (D+) results in the recipient lacking preformed anti-EBV cytotoxic immunity, which, upon the occurrence of primary EBV infection, facilitates the development of PTLD. 14 In the context of allo-HSCT, T-cell depletion, whether conducted ex vivo or in vivo, significantly influences the risk of PTLD. The use of ATG, particularly in transplants involving matched unrelated donors, markedly increases PTLD incidence. 15 This effect is similar to that observed with ex vivo T-cell depletion protocols, such as those utilizing anti-CD52 alemtuzumab. 14 According to the study by Fujimoto et al., our patient was classified as being at intermediate risk for the development of PTLD, with a 4.6% likelihood of occurrence over a 2-year period. This study introduced a novel 5-point scoring system that incorporates three pretransplant risk factors to assess the risk of PTLD prior to allogeneic HSCT. These factors included the use of ATG in the conditioning regimen (high dose, 2 points; low dose, 1 point), donor type (HLA-mismatched-related donor, 1 point; unrelated donor, 1 point; cord blood, 2 points), and the presence of aplastic anemia (1 point). Based on the total score, patients were categorized into four risk groups: low risk (0 or 1 point), intermediate risk (2 points), high risk (3 points), and very high risk (4 or 5 points), with corresponding 2-year probabilities of 0.3%, 1.3%, 4.6%, and 11.5%, respectively. 16
Determining an optimal treatment protocol for T-cell and NK/T-cell PTLDs remains challenging. The initial therapeutic approach for these rare PTLD subtypes typically involves reducing immunosuppression. Unlike B-cell PTLD, where monoclonal antibody therapy has become the standard of care, T-cell and NK/T-cell PTLD often necessitate aggressive treatment strategies, including intensive chemotherapy, radiotherapy, and surgical intervention. 17 Matsumura et al. reported a case of late-onset multiple NK/T-cell PTLD in which CR was attained following surgical intervention combined with a reduction in immunosuppression. 18 Similarly, Acar et al. 19 presented a case of ENKTL, nasal type, occurring 14 years after renal transplantation, which was successfully managed through immunosuppression reduction and administration of the EPOCH chemotherapy protocol. In the present case, immunosuppression was reduced through CSA cessation and methylprednisolone tapering immediately following confirmation of NK/T cell PTLD. As a candidate for the CHOP chemotherapy regimen, only vincristine was administered before the patient’s clinical status worsened, leading to the discontinuation of the chemotherapy.
Conclusion
In summary, the case described here is a rare instance of early onset, EBV-positive NK/T-cell PTLD manifested as ENKL with hepatic lesions. Additional research is needed to clarify the risk factors and identify the most effective treatment approaches for NK/T-cell PTLD following allogeneic HSCT.
Footnotes
Acknowledgements
We are thankful for the help of our co-workers at our institution in the clinical management of this patient.
Ethical Considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Consent to Participate
Written informed consent was obtained from a legally authorized representative for anonymized patient information to be published in this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All available data are presented in this article. If further explanation is required, please contact the corresponding author.