
Abstract
Schnitzler’s Syndrome (SS) is a rare late-onset acquired autoinflammatory disorder which consists of chronic urticaria associated with a monoclonal IgM-kappa gammopathy, arthralgias, skeletal hyperostosis, lymphadenopathy, and recurrent constitutional symptoms. The average age of diagnosis is 51 years with a slight male predominance with a male to female ratio of 1.6. Diagnosis of SS requires the presence of 2 major criteria including chronic urticaria and monoclonal IgM along with at least two of the following minor criteria: recurrent intermittent fevers, bone pain, arthralgias, elevated erythrocyte sedimentation rate (ESR), neutrophilic dermal infiltrate on skin biopsy, and leukocytosis or elevated C-reactive protein (CRP). Early diagnosis and clinical awareness are paramount in SS as it is associated with a 15-20% risk of lymphoproliferative malignancy. The median overall survival is 12.8 years. We present a case of a 39-year-old female with new onset urticaria associated with recurrent fevers and joint pain. Symptoms were refractory to steroids, and high dose antihistamines. Multi-disciplinary evaluation resulted in the ultimate diagnosis of Schnitzler’s Syndrome. The patient was ultimately treated with canakinumab (Il-1 inhibitor), with near resolution of symptoms. This case demonstrates the importance of a broad differential diagnosis and maintaining a high clinical suspicion for rare diseases when presented with a complex form of an otherwise common condition.
Introduction
Schnitzler’s Syndrome (SS) is a rare late-onset acquired autoinflammatory disorder which consists of chronic urticaria associated with a monoclonal IgM-kappa gammopathy, arthralgias, skeletal hyperostosis, lymphadenopathy, and recurrent constitutional symptoms. 1 The average age of diagnosis is 51 years with a slight male predominance and median overall survival of 12.8 years.1,2 There is not one specific test for SS and therefore this auto-inflammatory syndrome is often underdiagnosed. 2 Early diagnosis of SS is often reliant on a high clinical index of suspicion and is paramount as it is associated with a 15-20% risk of lymphoproliferative malignancy similar to those who hold a diagnosis of monoclonal IgM gammopathies of undetermined significance (MGUS). 3 Treatment with IL-1 inhibitors such as anakinra, canakinumab, and rilonacept have been associated with symptom resolution and in cases achievement of remission. 4 While the exact pathophysiology of SS is not well understood, the diagnosis is ultimately made when two major criteria of urticarial skin rash and monoclonal IgM component are met along with at least two of the following: recurrent intermittent fevers, bone pain, arthralgias, elevated erythrocyte sedimentation rate (ESR), neutrophilic dermal infiltrate on skin biopsy, and leukocytosis or elevated C-reactive protein (CRP). 1
We present a case of a 39-year-old female with new onset neutrophilic dermatosis associated with recurrent fevers and joint pain. The patient had a complicated clinical course with an ultimate diagnosis of Schnitzler’s Syndrome.
Case Presentation
A 39-year-old female with a past medical history of endometriosis and generalized anxiety disorder initially presented to primary care with a 1-month history of bilateral upper extremity and lower extremity urticaria (Figures 1 and 2). Lesions were characterized as pruritic hives, with each lesion lasting greater than 24 hours and with residual hyperpigmentation following resolution. These symptoms were associated with a 10-year history of intermittent low-grade fevers and night sweats which had previously been worked up without identification of a clear etiology.

Anterior-shin Urticaria.

Left calf Urticaria.
She received oral steroids with initial improvement in urticaria but with recurrence upon tapering and discontinuation. She was evaluated by Dermatology who completed a punch biopsy of the skin lesion. Biopsy revealed neutrophils and perivascular lymphocytic infiltrates consistent with urticaria without vasculitis (Figure 3). The patient was then seen by Allergy and Immunology and placed on antihistamines. Initial dose was cetirizine 10 mg once to twice a day and was increased to a maximum dose of 20 mg twice daily without improvement in symptoms. Omalizumab, a monoclonal antibody targeting human immunoglobulin (Ig)E, was then considered for chronic urticaria. However, based on the patient’s systemic symptoms and a high clinical suspicion for a secondary cause of urticaria, the patient was referred to rheumatology prior to the initiation of omalizumab.

(A) (40×): Punch biopsy with mild perivascular inflammation. (B) (100×): Perivascular inflammation composed of lymphocytes and eosinophils. (C) (200×): Perivascular inflammation composed of lymphocytes and eosinophils.
At the time of evaluation by Rheumatology, the patient was also experiencing arthralgias specifically of the knees and pain in the anterior shin during urticarial flares. She also reported intermittent low-grade fevers and drenching night sweats. She had been on prednisone 5 to 10 mg without complete resolution of symptoms, requiring escalating steroids to high doses and missing multiple days of work.
The patient underwent an extensive autoimmune, infectious, and hematological workup (Table 1). She was noted to have an elevated CRP of 33.4 mg/L (normal: <3mL/L) and leukocytosis of 13.3 thou/cumm (normal: 4.5-11 thou/cumm). ANA, ANCA, dsDNA, CCP, and rheumatoid factor titers were unremarkable along with a negative anti-C1q, normal C1q, normal C3 and C4, and a normal complete metabolic panel. Infectious workups included Lyme IgG and IgM Ab, T Pallidum Ab, RPR, Malaria, HIV (human immunodeficiency virus) 1/ 2 Antigen/Ab, hepatitis A IgM, Hepatitis B Core Antibody IgM, Hepatitis B Surface Antigen and HCV (hepatitis C virus) AB were unremarkable.
Autoimmune, Infectious, and Hematological Workup.
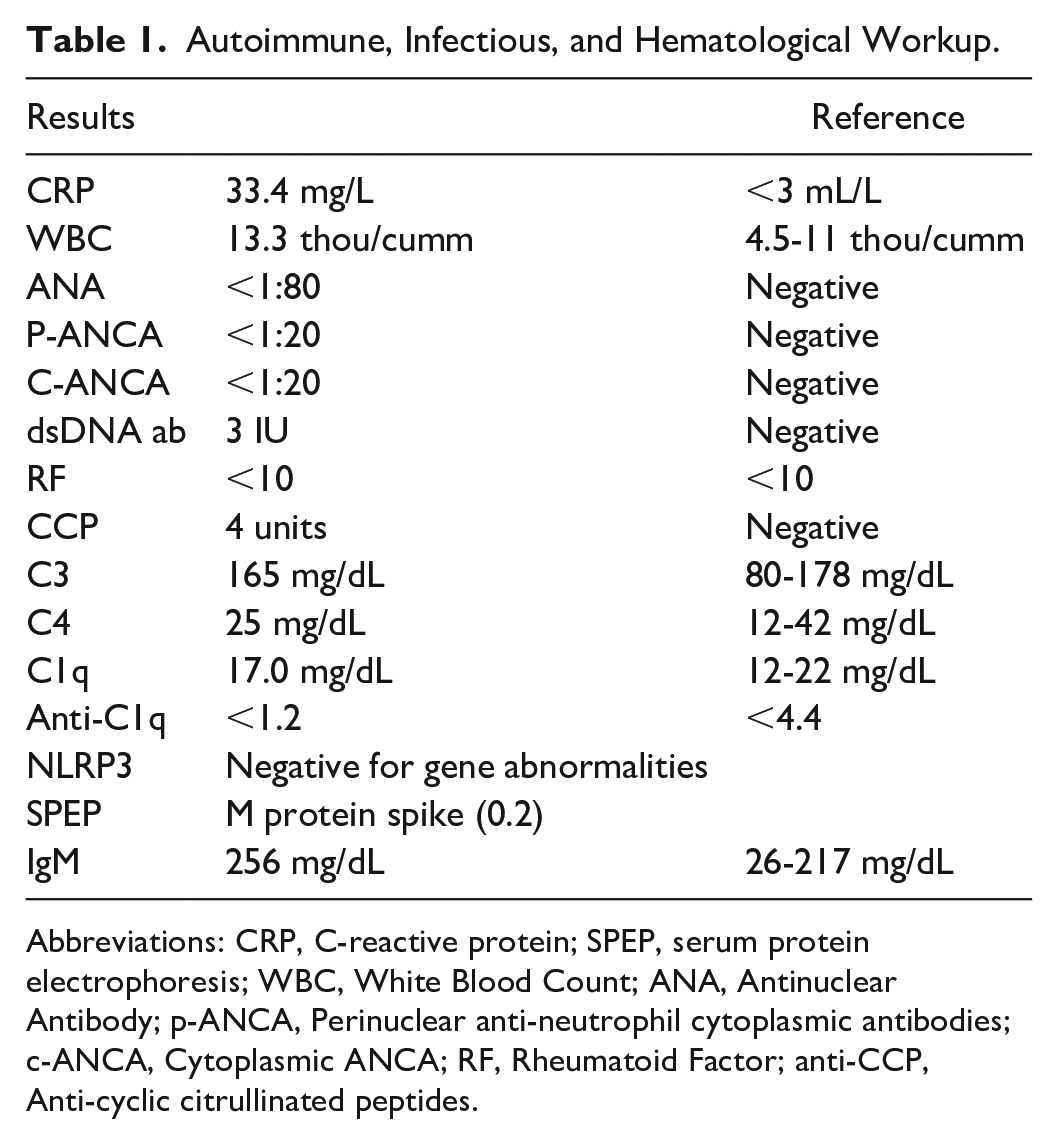
Abbreviations: CRP, C-reactive protein; SPEP, serum protein electrophoresis; WBC, White Blood Count; ANA, Antinuclear Antibody; p-ANCA, Perinuclear anti-neutrophil cytoplasmic antibodies; c-ANCA, Cytoplasmic ANCA; RF, Rheumatoid Factor; anti-CCP, Anti-cyclic citrullinated peptides.
Serum protein electrophoresis (SPEP) demonstrated an M protein spike (0.2) and serum IgM elevation at 255 mg/dL (normal: 40-245 mg/dL). Immunofixation showed IgM monoclonal protein with kappa light chain specificity. Given our patient’s persistent leukocytosis, a peripheral smear was examined, and smudge cells/atypical lymphocytes were seen concerning a mature B cell neoplasm. Peripheral flow cytometry redemonstrated the known mild leukocytosis however no overt evidence of leukemia or lymphoma was noted. Bone marrow biopsy and aspiration were also negative for lymphoma. Computed tomography (CT) of the chest, abdomen, and pelvis revealed no focal lesions or enhancements concerning for malignancy. X-rays of the hands, knees (Figure 4), shins, and bilateral hips showed no erosions, effusion, nor other findings concerning for an inflammatory process.

X-rays of bilateral knees.
An Invitae Autoinflammatory and Autoimmunity Syndromes Panel was performed and demonstrated a variable of uncertain significance (NFKB1); however, negative for NLRP3 gene abnormalities which excluded a diagnosis of cryopyrin associated periodic syndrome (CAPS). 5
After a multidisciplinary evaluation, Schnitzler’s Syndrome was identified as the diagnosis for this patient. Our patient met diagnostic criteria for this syndrome based on the presence of chronic urticaria, monoclonal IgM Kappa (both major criteria for SS). In addition to all minor criteria including recurrent intermittent fevers, bone pain, arthralgias, elevated ESR, neutrophilic dermal infiltrate on skin biopsy, leukocytosis and elevated CRP.
Prior to the diagnosis of SS, our patient’s symptoms had been recurrent requiring escalating glucocorticoid doses. Symptoms were also refractory to high dose antihistamine therapy, and she subsequently experienced debilitating paresthesias in lower extremities, worse during flare ups. After the diagnosis of SS, Anakinra, an interleukin-1 receptor antagonist, was prescribed but denied by her insurance company. As such, Dapsone 50-75mg daily was tried for approximately 6 weeks without improvement and with GI intolerance to the higher doses. Colchicine 0.6 mg twice daily was tried for 5 weeks without improvement. Finally, Canakinumab 150 mg q8 weeks was approved by insurance and initiated with a rapid and dramatic improvement in her symptoms. Unfortunately, she developed breakthrough symptoms at week 7, when she experienced a severe flare up. As such, the interval of Canakinumab was decreased to every 6 weeks with persistent symptomatic response and achievement of remission.
Discussion
Schnitzler’s syndrome is a rare auto-inflammatory disorder which shares clinical overlap with more common autoimmune, hematological, and immunological disorders leading to a delay of diagnosis by approximately 5 years. 1 While the exact pathogenesis of Schnitzler syndrome is unclear, the current hypothesis is thought to involve overproduction of IL-1 beta and IL-6. 6 A less understood association is the correlation between IL-1 production and clonal IgM production which is thought to be present in all patients diagnosed with Schnitzler syndrome. In addition to the IgM gammopathy, the presence of chronic urticaria with neutrophilic urticarial dermatosis on biopsy is another hallmark feature and major criteria in the diagnosis of SS. 7
The presentation of our patient with urticaria has a broad differential diagnosis. This includes chronic idiopathic urticaria, urticarial vasculitis, or other autoinflammatory syndromes. At first, our patient’s initial presentation of recurrent urticarial lesions was concerning for chronic idiopathic urticaria. Common urticaria is usually associated with flat erythematous plaques. On biopsy, common urticaria is not associated with an intense neutrophilic dermatosis of blood vessels and the interstitium will not show evidence of dermal edema or vasculitis. 8 It is important to note that the skin lesions in patients with SS are commonly described as slightly elevated plaques or red macules. 8 As noted above, skin biopsy of a patient with SS will show neutrophils and perivascular lymphocytic infiltrates consistent with urticaria without vasculitis. Ultimately, the presence of systemic symptoms, secondary hyperpigmentation, and monoclonal IgM-kappa gammopathy made a secondary cause of our symptoms more likely. Punch biopsy results without evidence of vasculitis, normal C1q, C3, and C4 also made hypocomplementemia urticarial vasculitis a less likely diagnosis.
Another diagnosis considered was cryopyrin associated periodic syndrome (CAPS) which is a family of autosomal dominant diseases, specifically, familial cold auto-inflammatory syndrome (FCAS), Muckle-Wells syndrome, and neonatal onset multi-systemic inflammatory disease (NOMID). These are thought to be caused by the gain of function point mutations in the NLRP3 gene. 5 Patients with CAPs present with episodic skin rashes, fevers, and joint pain. Clinical presentation makes it difficult to differentiate between SS. One key differentiator however is that patients with CAPS typically present in childhood years and do not have an IgM Gammopathy. Histologic examination of skin biopsy in patients with CAPS will also reveal perivascular neutrophilic dermal infiltrates in the dermis. 9 Patients with CAPS are spared from the 15% to 20% risk of lymphoproliferative malignancy that patients with SS are at risk for if left untreated. The incidence of reactive (AA) amyloidosis in patients with CAPS is up to 33% if untreated and even higher in other hereditary periodic fever syndromes. 10 The incidence of amyloidosis in SS is not well studied, but recorded incidence in is rare. 4
One diagnosis that is often initially considered by the general internist before SS is adult-onset Still disease (AOSD). The overall annual incidence of AOSD was 0.62/100 000 and overall prevalence is 6.77/100 000. 11 A hallmark feature of Adult Onset Still is a quotidian or double-quotidian fever with wide variance throughout the day which is not typically seen in SS. AOSD is associated with a non-pruritic maculopapular rash without urticarial features typically on the trunk and upper and lower extremities and presents during febrile episodes. On skin biopsy, deposition of C3 in the blood vessel walls are expected along with dermal edema and lymphocytic and histiocytic infiltration of the superficial dermis. 12 While a leukocytosis can be seen in both patients with SS and AOSD, one differentiating finding is that patients with AOSD are not expected to have IgM gammopathy.
Treatment of SS is known to be challenging largely due to the unclear pathophysiology that drives the disease and due to the absence of an Food and Drug Administration (FDA)-approved medication for this entity, which complicates insurance coverage. Similar to our case, it is not uncommon for symptoms to be refractory to steroids and high doses of antihistamine. It is becoming more widely accepted to utilize an IL-1 inhibitor such as anakinra, canakinumab, and rilonacept with very good response to treatment.1,13,14 The use of these medications has been associated with symptom resolution and, as in our case, with complete remission.
Summary
A multidisciplinary evaluation and discussion is needed in patients with atypical presentations of urticarial syndrome. When evaluating patients with chronic urticaria, joint pain and fevers, it is important for the general internist to pay attention to unusual symptoms, such as lesions lasting more than 24 hours and leaving hyperpigmentation or scaring. Ordering an appropriate autoimmune, hematological, and immunological testing while maintaining a high index of suspicion is important for the diagnosis of a rare auto-inflammatory disorder such as SS. Maintaining a broad differential diagnosis can result in early diagnosis and treatment which can dramatically improve a patient’s overall quality of life and long-term risk of malignancy.
Footnotes
Acknowledgements
The authors thank Dr Macartney Welborn and Dr Vladamir Vincek at the University of Florida Department of Pathology for providing punch biopsy figures.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Verbal informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
Prior Presentation of Abstract Statement
Poster Presentation- American Academy of Allergy Asthma & Immunology Annual Meeting- San Antonio, Tx 2/24/2023
Poster Presentation-Congress of Clinical Rheumatology—Destin FL 5/6/2023