
Abstract
Thrombotic thrombocytopenic purpura (TTP) is a potentially fatal condition that can be challenging for clinicians to identify in the setting of autoimmune diseases such as systemic lupus erythematosus (SLE). This difficulty is compounded when a patient presents with all of the clinical signs of a TTP-like microangiopathy, however, with near normal ADAMTS13. This case report describes a 44-year-old female with a history of SLE who was hospitalized with acute on chronic anemia, thrombocytopenia, and altered mental status. The patient’s ADAMTS13 was mildly low; hence, she was initially treated for SLE-associated immune thrombocytopenic purpura without any clinical response. The patient then underwent plasmapheresis (plasma exchange [PLEX]) for treatment of a suspected TTP-like microangiopathy. She responded to PLEX with improvement in her platelet count and mental status. This case illustrates the importance of considering TTP-like microangiopathic hemolytic anemia in the differential for patients with a history of SLE presenting with clinical signs of TTP even in the setting of near-normal ADAMTS13, thus warranting prompt treatment with PLEX.
Keywords
Introduction
Thrombotic thrombocytopenic purpura (TTP) is a potentially fatal condition that can complicate autoimmune diseases requiring urgent lifesaving treatment. However, early identification of this condition can be challenging for clinicians as there are increasing reports of patients with systemic lupus erythematosus (SLE) developing all of the classic signs of TTP, but without the low ADAMTS13. Here we present a case of a 44-year-old female who was hospitalized with a TTP-like condition without low ADAMTS13 and with improvement in her symptoms upon treatment with plasma exchange (PLEX).
Case Report
A 44-year-old female with past medical history of SLE and mechanical mitral valve replacement presented to the hospital with abdominal pain and black stools. She was prescribed mycophenolate mofetil, prednisone, and azathioprine for management of her SLE as an outpatient; however, the patient was only taking azathioprine due to issues with her insurance.
On presentation, the patient was found to have pancytopenia with platelets of 67 000 and acute renal failure (creatinine 3.0). She underwent a computed tomography (CT) scan of the abdomen and pelvis in addition to esophagogastroduodenoscopy (EGD). The patient’s CT scan showed a thickened gallbladder, which required no surgical intervention according to general surgery. Meanwhile, the EGD only revealed small erosions in her stomach. Rheumatology workup (Table 1) was consistent with SLE flare as demonstrated by elevated dsDNA and low C3/C4. She was initiated on intravenous methylprednisolone for 3 days to treat for SLE flare. Her thrombocytopenia temporarily improved with the corticosteroids; however, her renal function continued to decline. Therefore, she underwent a renal biopsy that revealed class IV lupus nephritis with crescentic glomerulonephritis (IFTA [interstitial fibrosis/tubular atrophy] 20% to 30%), but it did not show any evidence of thrombotic microangiopathy (TMA) or features of ANCA-associated vasculitis. The patient was subsequently initiated on mycophenolate-mofetil and prednisone for treatment of lupus nephritis. She was transitioned from warfarin to a heparin drip during the biopsy for management of her mechanical valve. In the meantime, her thrombocytopenia continued to worsen to a platelet count of 11 000.
Rheumatology Evaluation.

Abbreviation: Ig, immunoglobulin.
Her hospital course was soon complicated by acute encephalopathy. CT scan of the head was negative for acute intracranial pathology. Laboratory workup for pancytopenia (Table 2) was unrevealing. Her low haptoglobin and high lactate dehydrogenase (LDH) are indicative of a hemolytic process, but are not markers for a specific etiology of hemolytic anemia, especially with a negative direct Coombs test. She was also noted to have high ferritin, high iron, low total iron-binding capacity, and high iron saturation, which may suggest underlying anemia of chronic inflammation but does not explain the acute drop in hemoglobin. Antiphospholipid syndrome was also considered, since the patient had positive antiphospholipid syndrome antibodies (Table 1); however, there were no signs of thrombosis on imaging including Doppler ultrasound of lower extremities. Last, while the patient was on heparin, her partial thromboplastin time and international normalized ratio were elevated but fibrinogen was normal with only mildly elevated d-dimer rendering disseminated intravascular coagulation less likely.
Pancytopenia Evaluation.
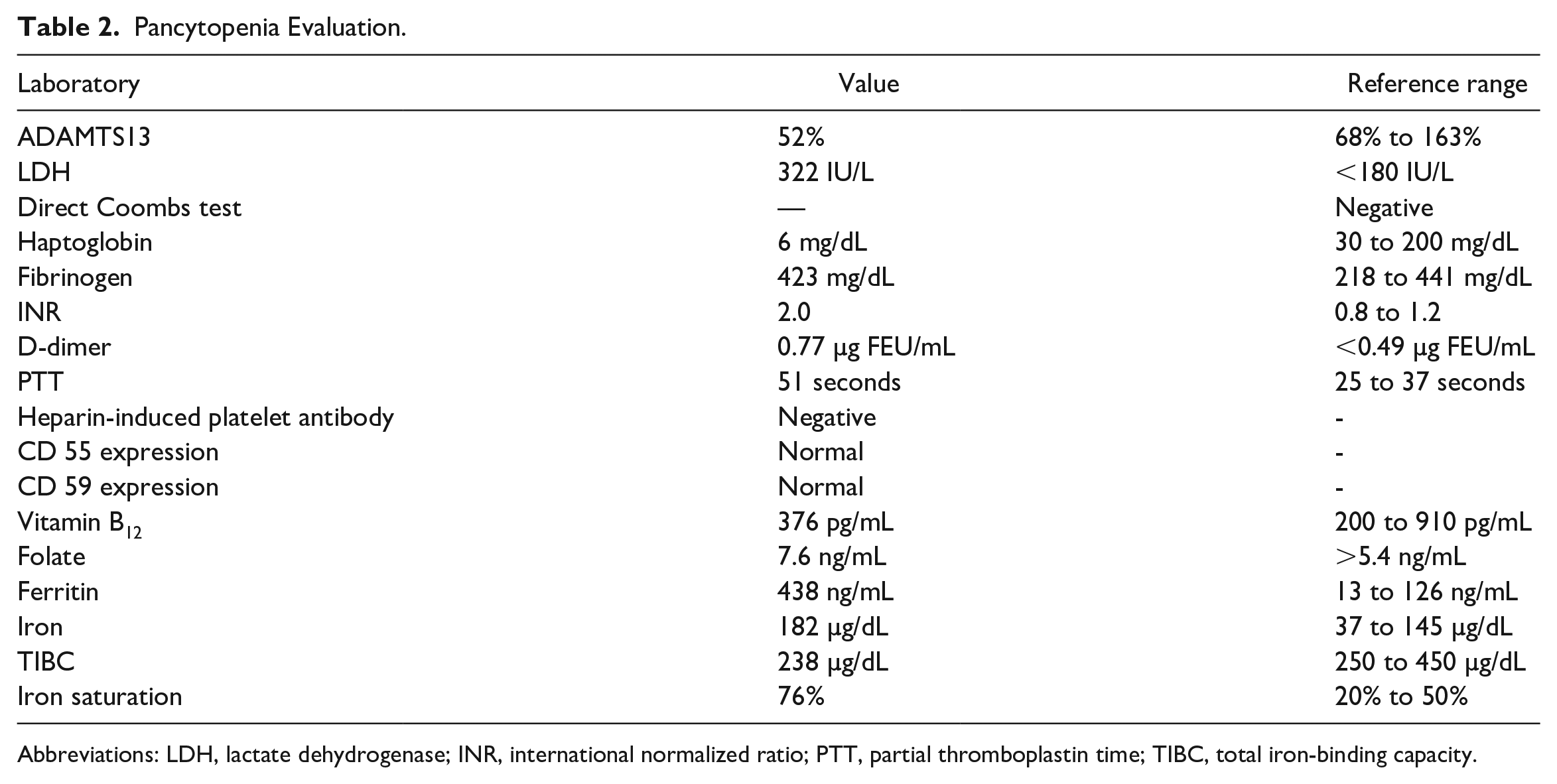
Abbreviations: LDH, lactate dehydrogenase; INR, international normalized ratio; PTT, partial thromboplastin time; TIBC, total iron-binding capacity.
Rheumatology and hematology were consulted, and produced a differential diagnosis for the patient’s thrombocytopenia that included TTP, complement-mediated hemolytic uremic syndrome (CM-HUS), SLE-associated immune thrombocytopenic purpura (ITP), heparin-induced thrombocytopenia (HIT), and drug-induced thrombocytopenia from the mycophenolate mofetil. It was unclear whether her mental status changes were from uremia or a condition such as TTP, steroid-induced, or lupus-associated flare affecting the central nervous system. HIT assay returned negative and ADAMTS13 returned at 52%, which was less suggestive for TTP. Peripheral smear revealed few to moderate schistocytes with burr cells (Figure 1); however, these schistocytes could be due to her mechanical valve rather than any active disease process. Furthermore, CM-HUS was less likely given that no TMA was found on renal biopsy. She was started on Rituximab in addition to another round of intravenous steroids to treat for possible SLE-associated ITP. Her renal function (creatinine 5.5) and azotemia (blood urea nitrogen 123) continued to worsen, which led to the patient being initiated on hemodialysis. She was transfused platelets without much response.

Peripheral smear showing few to moderate schistocytes and burr cells.
Although we initially thought TTP was less likely in the setting of a normal ADAMTS13, the treatments for ITP were unsuccessful, HIT assay returned negative, and she had been on mycophenolate mofetil without any drug-induced thrombocytopenia in the past. Thus, the patient was treated preemptively with PLEX for a TTP-like microangiopathy associated with her SLE. Her mycophenolate mofetil was held simultaneously in an attempt to rule out drug-induced thrombocytopenia from the differential. This resulted in an immediate improvement in her platelets, and her LDH normalized by the second session of PLEX. Mycophenolate mofetil was reinitiated because we reasoned that if this was drug induced from bone marrow suppression, such a stark improvement would not be seen over the course of a day. The patient continued to receive hemodialysis and PLEX on alternate days with improvement of platelets to >100 000 after 6 PLEX treatments. Her renal function improved during the hospital course to the point where she no longer required hemodialysis.
The patient underwent bone marrow biopsy to rule out any other potential causes for her pancytopenia. Her bone marrow biopsy showed cellular marrow with trilinear hematopoiesis, blasts <5%, and no evidence of acute leukemia, lymphoma, myelodysplasia, monoclonal B-cell population, or ring sideroblasts. The lymphoid cells were predominantly T-cells with a CD4:CD8 ratio of 1.27:1. No monoclonal surface light chain expression was demonstrated to suggest B-cell lymphoma. The kappa:lambda ratio was 1.78:1. The B cells were negative for CD5 and CD10. We concluded that her clinical course was consistent with SLE-associated TTP-like microangiopathic hemolytic anemia (MAHA) without a very low ADAMTS13.1,2
Discussion
Systemic lupus erythematosus is a complex autoimmune disease that can manifest as mild symptoms to life-threatening flares. Thrombocytopenia is one of the more common manifestations in SLE affecting 20% to 40% of patients and is one of the criteria for SLE as presented by the American College of Rheumatology.3,4 The most common cause of thrombocytopenia in these patients is the development of autoantibodies to platelets, also recognized as SLE-associated ITP. Studies have shown that SLE patients with thrombocytopenia have elevated levels of platelet-binding immunoglobulin G and platelet-associated immunoglobulin G. 5 Unlike isolated ITP, these autoantibodies do not bind to platelet glycoproteins. Most of the time, thrombocytopenia in SLE falls into 2 groups: acute or chronic. Usually acute thrombocytopenia is easier to manage than chronic thrombocytopenia. 6 High-dose glucocorticoids are utilized as first-line therapy; however, for thrombocytopenia that does not respond to steroids, other immunosuppressive agents can be used such as Rituximab. 3 Thrombocytopenia in SLE has been strongly associated with other manifestations of SLE such as neuropsychiatric disease, hemolytic disease, and renal disease. 3 Thus, it was initially thought that our patient’s altered mental status could have been a manifestation of an SLE disease flare in conjunction with a phase of acute thrombocytopenia. However, our patient did not respond to high-dose steroids nor Rituximab.
Not as common, TTP can be another contributing phenomenon in SLE patients with thrombocytopenia. Unlike SLE-associated ITP, TTP is a life-threatening TMA that is typically defined by a low von Willebrand factor cleaving proteinase called ADAMTS13. Levels of <10% are diagnostic. 7 However, there is evidence of acquired TTP that can be caused by development of von Willebrand multimers that overwhelm a patient with normal levels of ADAMTS13. 8 TTP occurs when platelet-rich microthrombi form in blood vessels contributing to overall thrombocytopenia, which then leads to anemia by shearing of erythrocytes along these microthrombi in the blood vessels. It has a mortality rate as high as 5% to 16% with almost half of the cases resulting in a relapse. 7 Thus, early identification and treatment is key. Several scores have been derived to help with clinical diagnosis of this deadly condition including the Bentley score and the PLASMIC score; however, these are only to guide clinicians.9,10 This condition can lead to ischemic-related end-organ damage from seating of thrombi in vessels. Schistocytes are often identified on peripheral smear, an indication of MAHA seen in TTP. 11 Patients may present with signs of visceral organ involvement with the central nervous system first to be affected. Signs of nervous system involvement can range anywhere from confusion to focal deficits consistent with stroke. Gastrointestinal symptoms and acute renal failure are also common. 7 Our patient had all 3 aforementioned clinical signs of TTP; however, her renal failure was more likely caused by active lupus nephritis. The treatment is emergent PLEX, which our patient received with improvement in platelet count and mental status.
Complement-mediated hemolytic uremic syndrome, also commonly known as atypical HUS, is another TMA that can present similarly to TTP. It is caused by uncontrolled activation of the alternate complement pathway, which can be genetic or acquired. The typical features of this condition include less severe thrombocytopenia (usually platelets >30 × 109/L), microangiopathic hemolytic anemia, and acute renal failure. 12 There is usually evidence of TMA on kidney biopsy as the root cause of the patient’s renal failure. 12 However, our patient’s kidney biopsy was more consistent with active lupus nephritis and did not show any TMA. Similarly, C3 and C4 were both low in the setting of elevated dsDNA antibodies, which was more consistent with a lupus flare than complement activation from the alternate complement pathway, which would typically lead to low C3 but normal C4 levels. 12 Our patient had elevated LDH, which is expected in MAHA; however, LDH levels in CM-HUS are typically >600 IU/L. 13 For these reasons, we diagnosed our patient with a TTP-like MAHA instead of CM-HUS. Nevertheless, in patients where PLEX does not improve or only temporarily improves thrombocytopenia, treatment for CM-HUS with eculizumab should be considered. 12
Thrombotic thrombocytopenic purpura and SLE flares have overlapping features adding to the complexity of discerning between the two. Contrary to the accepted diagnostic criteria for TTP, our patient had ADAMTS13 levels of 52% which is mildly low. However, there are documented cases of patients with mildly low to normal ADAMTS13 but developing a TTP-like condition via the mechanism proposed in Franchini et al 8 described above. Given the wide number of similar presentations, Blum and Blake have coined the term SLE-associated TTP-like MAHA. 2 A study conducted in Japan showed that TTP in patients with connective tissue diseases generally had higher ADAMTS13 levels than in idiopathic TTP thus proposing different pathophysiology behind SLE-associated TTP and idiopathic TTP. 14 All of which supports this patient having TTP-like MAHA induced by the patient’s SLE.
Conclusion
The significance of this case report is to press the importance of considering TTP-like MAHA in the differential for patients with a history of SLE presenting with anemia, thrombocytopenia, and other clinical signs of TTP. Additionally, ADAMTS13 may be mildly low to normal in these patients making it an easily missed diagnosis and easily confused for CM-HUS. As this is a potentially life-threatening illness, there should be a low threshold for initiating PLEX in patients with a clinical course consistent with SLE-associated TTP-like MAHA.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.