
Abstract
Wolfram syndrome (WS) is a rare genetic disorder typically characterized by juvenile onset diabetes mellitus, optic atrophy, hearing loss, diabetes insipidus, and neurodegeneration. There would be a high index of clinical suspicion for WS when clinical manifestations of type 1 diabetes and optic atrophy present together. Genetic analysis is often required to confirm the diagnosis. We describe a pair of Chinese siblings diagnosed with WS at ages 20 and 24 years, respectively. DNA sequencing of the WFS1 gene which encodes for Wolframin ER Transmembrane Glycoprotein identified a heterozygous nonsense variant NM_006005.3: c.1999C>T p.(Gln667*) and a heterozygous missense variant c.2170C>T p.(Pro724Ser) in exon 8 of the gene for both siblings. There is no curative treatment for WS and management of this debilitating disease is aimed at treating individual clinical manifestations, slowing disease progression, and improving quality of life. Treatment with liraglutide, a glucagon-like-peptide-1 receptor agonist, and tauroursodeoxycholic acid was started for the younger sibling, the proband. There was reduction in insulin requirements and improvement in glycemic control. The other sibling was not offered liraglutide due to her complex treatment regimen for end-organ failure. Genetic testing is a valuable tool to detect WS early to allow precise and prompt diagnosis, thereby facilitating the coordinated care from a multidisciplinary team of clinicians.
Introduction
Wolfram syndrome (WS) is a rare genetic disorder, characterized by juvenile onset diabetes, optic nerve atrophy leading to progressive vision impairment, and neurodegeneration.1,2 Other clinical manifestations include, but are not limited to, diabetes insipidus, hearing loss, and neurogenic bladder. These may vary along a clinical spectrum from mild to severe.2,3 The estimated prevalence varies widely, from 1 in 100 000 in North America, 4 1 in 351 000 in Italy, 5 1 in 710 000 in Japan, 6 1 in 770 000 in the United Kingdom, 1 to 1 in 805 000 in North India. 7 The prevalence elsewhere is unknown.
Due to the rarity of this disorder, it can be easily mistaken for typical type 1 diabetes before other symptoms such as optic atrophy manifest, hence it can be challenging for clinicians to obtain an early diagnosis without a high index of suspicion. Genetic analysis is often required to confirm the diagnosis.
Herein, we report a family of 2 siblings who were diagnosed relatively late with WS at ages 20 and 24 years, respectively.
Case Presentation
Case 1—Proband
A 19-year-old Chinese man, believed to have type 1 diabetes from 6 years of age, was transitioned to the adult endocrinology service. He had a history of asymptomatic atrial septal defect of 5 mm, monitored regularly by a cardiologist. He did not smoke or drink alcohol. He had an elder sister who also had type 1 diabetes but no other significant family history. Glutamic acid decarboxylase antibodies, islet cell antibodies, or C-peptide level were not available. He was switched from multiple daily insulin injections to continuous subcutaneous insulin infusion (CSII) therapy which improved his glycemic control (glycated hemoglobin, HbA1c from 10% to 8%). He weighed 46.2 kg, height 1.64 m, with a body mass index of 17.2 kg/m2.
He reported gradual blurring of vision, was referred to ophthalmology, and found to have bilateral disk pallor without evidence of diabetic retinopathy. His last ophthalmologic screening with the pediatric service more than a year prior was normal. Further history revealed that his sister had optic disk atrophy with severe retinopathy, which was attributed to poor diabetes control. He had a previous traumatic head injury 4 years prior which resulted in left parietal fracture and right temporal hematoma. Computed tomography of the brain did not reveal any intracranial space-occupying lesions. Subsequent magnetic resonance imaging (MRI) orbits did not identify dorsal midbrain lesions. There was no enhancing mass in the bilateral orbits and the optic nerve and optic chiasm appeared unremarkable with no mention of cerebral atrophy. Syphilis screen was negative.
Based on the features of juvenile onset diabetes and optic disk pallor, Wolfram Syndrome (WFS1; MIM 222300) was suspected and consent was obtained for genetic analysis. DNA sequencing of the WFS1 gene, which encodes for Wolframin ER Transmembrane Glycoprotein, identified a heterozygous nonsense variant NM_006005.3: c.1999C>T p.(Gln667*) and a heterozygous missense variant c.2170C>T p.(Pro724Ser) in exon 8 of the gene (Figure 1). Both variants are present in the general East Asian population (Genome Aggregation Database [gnomAD]) at an extremely low frequency (<0.1%) and were reported as pathogenic variants in ClinVar. Notably, there were several cases8-10 reported with WS who were compound heterozygous for the WFS1 p.(Gln667*) variant and distinct frameshift variants in the same gene. Genetic testing for first-degree relatives was also performed. We identified the same compound heterozygous variants in the sister who had diabetes (case 2), while the parents and another brother who are healthy are carriers for either one of the variants (Figure 2). Results of the genetic testing were informed to the proband and the family members with genetic counseling provided. There was no consanguinity.
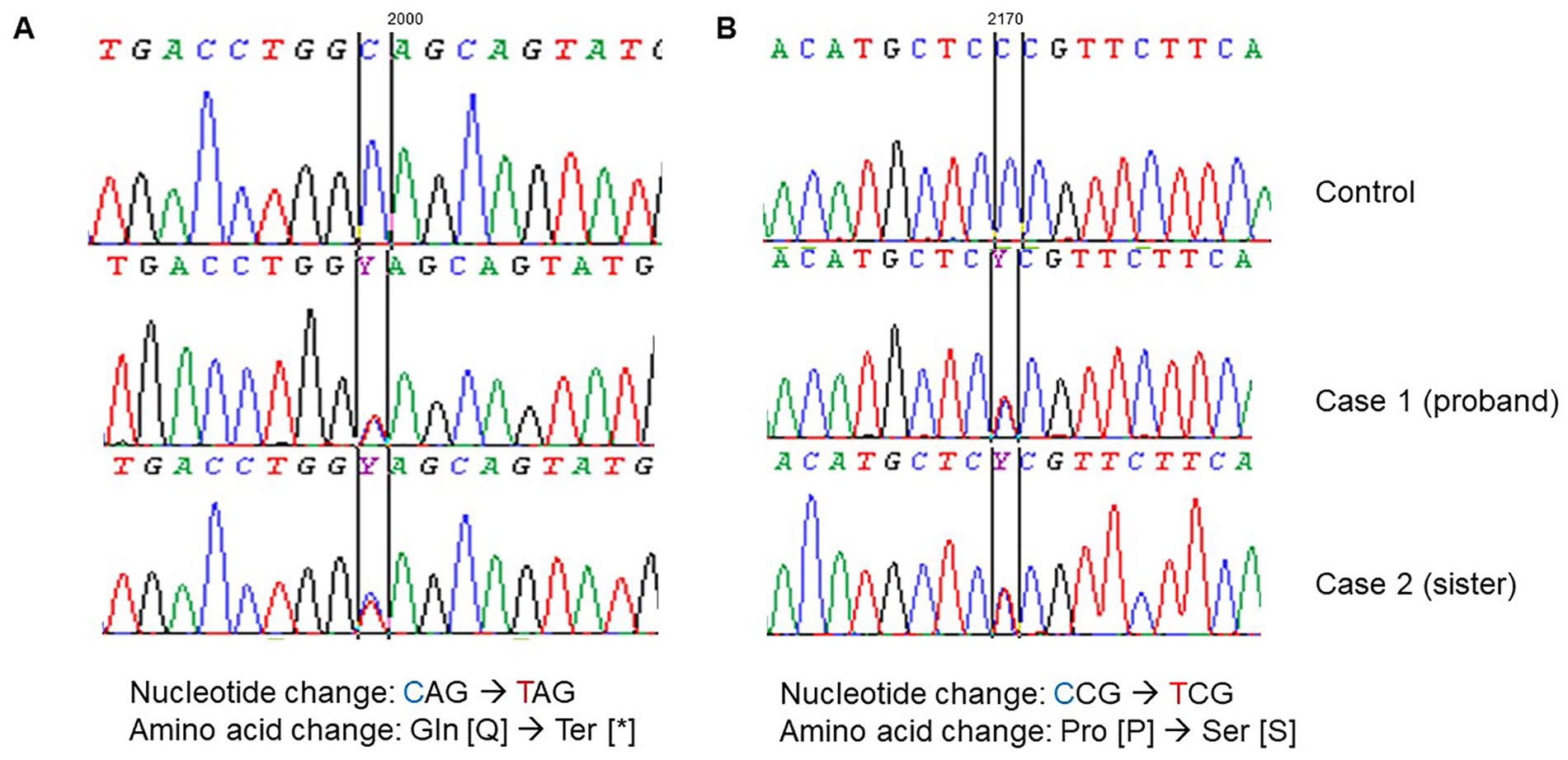
DNA sequencing of the WFS1 gene identified compound heterozygous variants in the proband and his sister: (A) c.1999C>T p.(Gln667*), (B) c.2170C>T p.(Pro724Ser). Both variants were identified using Sanger bidirectional sequencing.

Family pedigree showing segregation of WFS1 variants. Both the proband (II.3) and his sister (II.1) are diagnosed with Wolfram syndrome. Their father, mother (I.1 and I.2, respectively), and brother (II.2) are not affected, but are carriers of either the WFS1 p.(Gln667*) or p.(Pro724Ser) variant.
Further workup was done to look for other clinical manifestations associated with WS and he was referred to a multidisciplinary team from neurology, urology, and otorhinolaryngology specialties. Renal ultrasound revealed normal-sized kidneys with normal echogenicity without hydronephrosis. The urinary bladder was partially distended. Pre-void urinary bladder volume was 244 mL and post-void residual volume was 167 mL. He was diagnosed to have neurogenic bladder. He had no prior urinary tract infections. His audiogram was normal and he did not have features of hypogonadism, diabetes insipidus, or psychiatric manifestations. Although neurological examination was normal, a repeat MRI brain 1 year after diagnosis showed age advanced global cerebral and cerebellar volume loss with bilateral optic nerve atrophy.
A trial of glucagon-like-peptide-1 receptor agonist (GLP1-RA) was offered to him. He was counseled that GLP1-RA was a potentially useful therapy in WS but conclusive results were as yet unknown. He was also counseled on the need to reduce his total daily insulin requirements in view of the addition of GLP1-RA, whereupon he received close monitoring of his blood glucose levels, reminders not to skip insulin injections, and sick day advice on diabetic ketoacidosis management. With the GLP1-RA, his insulin requirements reduced by 20% and his HbA1c improved further to 7%. He was also started on tauroursodeoxycholic acid (TUDCA).
Case 2
Our second case is a 24-year-old woman, the elder sister of the proband who has the same compound heterozygous variants for WS. She was diagnosed with type 1 diabetes from the age of 3. At 21 years old, during her first visit to the center, her HbA1c was consistently >14% for several years with a history of poor adherence to insulin therapy and follow-ups, as well as previous admissions for diabetic ketoacidosis. She had diabetes-related complications of peripheral neuropathy, proteinuria, and features of retinopathy in bilateral eyes requiring photocoagulation. Two years later, she commenced CSII, a year after her younger brother, having seen the benefits in her sibling, and HbA1c improved to 8% to 10%. She was a nonsmoker and did not drink alcohol. She weighed 42.5 kg, height 1.50 m, with a body mass index of 18.8 kg/m2.
She had multiple episodes of lower urinary tract infections and pyelonephritis, resulting in diabetic ketoacidosis and hospitalizations. Renal ultrasound performed during her hospitalizations for urinary tract infections showed slight increased echogenicity of the kidneys, indicative of renal parenchymal disease but did not reveal any urinary calculi or hydronephrosis. Her care was complicated by issues of treatment refusal and lack of adherence to medical recommendations. She was later diagnosed with neurogenic bladder with high post-void residual urinary volume, necessitating intermittent self-catheterization. In the same year, routine ophthalmology review noted optic pallor in the left eye and proliferative retinopathy in bilateral eyes.
It was at this point that her younger brother had been confirmed with WS and genetic testing was also offered to her. By 24-years-old, she had both autonomic and peripheral neuropathy with neuropathic pain in her feet and gastroparesis noted on gastric emptying study. She had complete blindness in the left eye, limited visual capacity in the right eye with previous retinal surgeries, and was initiated on renal replacement therapy for end-stage renal failure. An MRI brain performed in the same year for first-onset focal seizure showed age-advanced global parenchymal volume loss, mild volume loss in the cerebellar hemispheres, pontine atrophy, and atrophy of the optic nerves and optic chiasm.
Between the ages of 18 and 23 years, she was sequentially diagnosed with eating disorder, suicide and self-harm attempts with insulin and multiple drug overdose, adjustment disorder, acute situational reaction with low mood and poor coping mechanism, and had often declined psychological assessments and interventions. She had no sensorineural hearing loss, ataxia, or clinical features of diabetes insipidus.
Discussion
WS was initially thought to be an autosomal recessive disease 11 resulting in dysfunctional and improperly folded endoplasmic reticulum (ER) wolframin protein. However, recent genetic analysis has found that a minority of patients carry the dominant WFS1 variants instead.12,13 The recessive presentation of WFS1 mutations continue to be termed as WS, whereas the dominant form of mutations is termed as Wolfram-like syndrome. 14 WS can also be further classified into type 1 (WS-1) and type 2 (WS-2) with WS-1 having variants (recessive or dominant mutations) in the WFS1 gene and WS-2 having CISD2 gene variants transmitted in an autosomal recessive mode. 15 We report a pair of siblings with compound heterozygous variants p.(Gln667*) and p.(Pro724Ser) in the WFS1 gene.
Due to diverse phenotypic expressions, variable age of clinical features, and different rate of disease progression, it is difficult to establish a clear genotype-phenotype correlation for this rare disease. 16 Diagnosis may be delayed due to this phenotype variability. It is now acknowledged that WS should be recognized as a spectrum disorder with mild to severe clinical manifestations.
Our first patient had initially presented with insulin-dependent diabetes at age 6, and later had decreased visual acuity at age 19, prompting the use of genetic tests to confirm the diagnosis at age 20, and involvement of a multidisciplinary team to monitor other clinical manifestations of WS. His symptoms are congruent to reports1,16 documenting the natural history of WS and median age of symptom onset. If it was not for the brother who had genetic testing for WS, the sister, who had multiple comorbidities which were attributed initially to long duration of suboptimal diabetes control, would not have been diagnosed with WS.
Although WS was commonly known as DIDMOAD, 1 an acronym for diabetes insipidus (DI), diabetes mellitus (DM), optic atrophy (OA), and deafness (D), one study showed that only 28% of patients displayed the full DIDMOAD phenotype. 16 In this same study of 392 patients, 16 the pattern of symptom onset is typically DM during first decade of life; OA, DI, and deafness in the second decade; urological issues and neurodegeneration in the third decade with a median age of death at 30 years, usually due to central respiratory failure with brainstem and cerebellar atrophy. 1 Our proband and sibling did not have sensorineural hearing loss or DI.
The timeline to reach diagnosis may be prolonged unless genetic testing is considered for those with clinical features suggestive of WS, 17 patients with type 1 diabetes who test negative for islet cell antibodies, 17 and usually one other manifestation such as optic atrophy. Although optic atrophy was the clinical feature that triggered our suspicion for WS in our proband and is the second most common clinical feature with an overall prevalence of 82%, 16 our 2 patients developed optic atrophy >10 years after diagnosis of diabetes. It will still be important that children with type 1 diabetes have an initial comprehensive ophthalmologic examination 2 to 5 years after diagnosis if they are aged ≥11 years or after puberty has started (whichever is earlier).18,19 Subsequent retinopathy screening should take place every 2 years, or more frequently, based on risk factor assessment, as per routine recommendations.18,19
Clinical Treatment Options
WS remains a debilitating and progressive disease with no effective treatment to halt or reverse its progression. The use of regenerative and gene therapy remains under the realms of research. 20 There are other treatment options proposed to manage WS, mainly targeted at reducing ER stress. These are not curative, but are aimed at treating individual clinical manifestation, slowing disease progression, and improving quality of life.
These treatment options include dantrolene, a muscle relaxant drug used for multiple sclerosis and cerebral palsy, targeting ryanodine receptor in the ER membrane preventing beta-cell death 21 ; dipeptidyl peptidase-4 inhibitor, a drug to treat diabetes which deactivates glucagon-like-peptide-1 hence providing protection against beta-cell failure 22 and possibly diabetes remission 23 ; valproate acid, an anti-epileptic drug reported to have neuroprotective function by inhibiting ER stress–induced apoptosis. 24
The first patient was initiated and maintained on liraglutide 0.6 mg per day, a GLP1-RA, upon the diagnosis of WS. The GLP1-RA has been used in patients with type 2 diabetes to stimulate beta-cell proliferation, augment insulin secretion, decrease glucagon production, and decrease beta-cell apoptosis. 25 Increasingly, GLP1-RA is also recommended as an adjunctive therapy to insulin for patients with type 1 diabetes. 26 A randomized controlled trial in Denmark 27 showed significant reduction in weight and insulin requirements but no improvement in HbA1c for normal-weight patients who have type 1 diabetes and poor beta-cell reserves. This differed slightly from our proband who had a HbA1c reduction of 8% to 7% with liraglutide.
For patients with WS, GLP1-RA is also a form of ER stress reducer used in animal models as a prophylactic treatment to delay the onset of diabetes and protect against vision loss. 28 Its anti-inflammatory and antioxidative properties have beneficial effects on the central and peripheral nervous systems 29 which could delay syndrome progression and potentially increase the number of healthy life years in individuals with WS. In addition to our case report, liraglutide was also reported to be safe and well tolerated in a case series of 4 pediatric patients with WS who were treated with it for 8 to 27 months. 30 There was no deterioration in C-peptide secretion, no significant changes in ophthalmological and neurological parameters, and no new WS clinical features during the follow-up. Nevertheless, larger clinical studies are needed to investigate the long-term effectiveness of GLP1-RA in the management of patients with WS.
While our proband received liraglutide after confirming the diagnosis of WS, the second patient, however, did not receive GLP1-RA due to her complex treatment regimens for multiple end-organ failure. The clinical team decided not to initiate a GLP1-RA due to limited clinical evidence of its use in patients on renal replacement therapy.
It has been reported that a combination treatment of chemical chaperones such as 4-phenylbutyric acid and TUDCA mitigated ER stress, 31 reduced mitochondrial dysfunction, 32 thereby preserving the function of beta cell and delaying hearing loss and neurodegeneration in patients with WS. 33 TUDCA was sourced online by the parents and provided to both patients.
Conclusion
The WS is now recognized as a spectrum disorder with variable combination and severity of clinical manifestations, making it difficult to diagnose and manage. The condition can be easily overlooked and underdiagnosed hence it is important to consider genetic testing when there are suspicious clinical features pointing toward WS. In addition, the use of GLP1-RA as a disease-modifying agent for WS may be considered while further clinical trials identify effective treatment options.
Footnotes
Acknowledgements
We would like to thank the patients for giving their consent to share their case for the benefit of improving care for individuals with Wolfram syndrome.
Author Contributions
D.P., E.Y., and C.T. drafted the manuscript and all authors read, revised and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Genetic analysis was supported by Khoo Teck Puat Hospital Science-Translational and Applied Research Grant 17201.
Ethical Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Verbal informed consent was obtained from the patients for their anonymized information to be published in this article.