
Abstract
Recent progress in laboratory techniques, particularly, identification of novel disease-causing genes, has led to the detection of different gene mutations that might be implicated in the pathogenesis of different hematological disorders like pure red cell aplasia (PRCA) and neutropenia. An autoinflammatory disorder known as deficiency of adenosine deaminase 2 (DADA2) has been recently noticed to present with variable hematologic abnormalities. We report 2 patients who presented with hematologic abnormalities in which 2 ADA2 gene mutations were detected. The first case is a 5-year-old girl who presented with severe PRCA and autoimmune hemolytic anemia without any other manifestation of DADA2 that resulted from a novel CECR1 c.714_738dup, p. (Ala247Glnfs*16) homozygous variant. The second case is a 10-year-old boy, known to have Hodgkin lymphoma and was under follow-up for 6 years; he presented with persistent neutropenia and was discovered to be homozygous for ADA2 c.1447_1451del, p. (Ser483Profs*5). In conclusion, we report two different novels ADA2 variants in two children; the first presented with PRCA and the second presented with persistent neutropenia. This report aims to raise the concerns regarding the use of genetic testing in different hematologic diseases with indefinite etiology, as it will lead to the best therapeutic strategies without the need for unnecessary interventions.
Introduction
Deficiency of adenosine deaminase 2 (DADA2) is an autoinflammatory disorder characterized by various forms of vasculitis and is rarely manifesting as pure red cell aplasia (PRCA) or neutropenia. Very few reports in the literature document these associations. We, therefore, report two different novel mutations in the ADA2 (also known as cat eye syndrome chromosome region 1 gene) in two different patients that result in DADA2 and presented with pure red cell aplasia in the first reported patient and persistent neutropenia in the second one. PRCA is normocytic normochromic anemia with severe reticulocytopenia and markedly reduced erythroid precursors in the bone marrow. It can be inherited as in Diamond-Blackfan anemia (DBA) or acquired. 1 The latter can be further classified into parvovirus-associated PRCA, drug-induced, primary idiopathic which is frequently antibody-mediated autoimmune disorder, or secondary PRCA that can be due to collagen vascular disorders, autoimmune diseases, lymphoproliferative disorders, or malignancies. 2 On the other hand, persistent neutropenia can result from different disorders such as malignancy, hematologic disorders, metabolic disorders, or infectious causes. Determining the cause of neutropenia is critical in defining a successful plan for treatment.
Case Presentation
Case 1
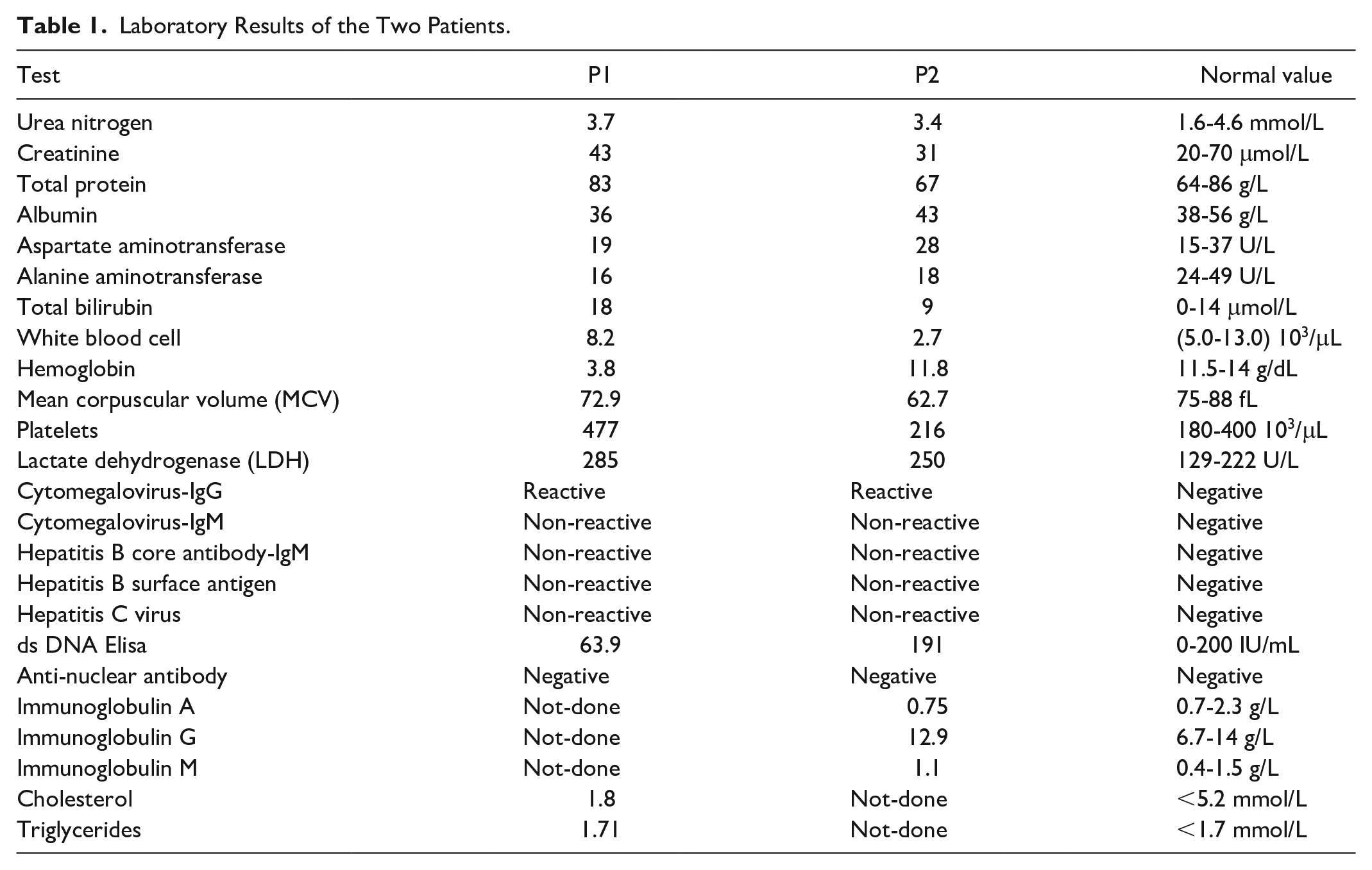
This is a 5-year-old Saudi girl who presented with generalized weakness, easy fatigability, pallor, and poor oral intake, O2 saturation 97%, and the patient was hemodynamically stable. She was admitted to the neonatal intensive care unit (NICU) at birth when her first blood count showed hemoglobin (Hb) of 4 gm/dL for which she received a blood transfusion. Initial investigation included a normal flow cytometry, that ruled out malignancy or immunologic disorders, and molecular genetics that confirmed a diagnosis of α-thalassemia trait which had no clinical implications. Further investigations were planned but the patient was lost to follow-up. For 5 years till the current presentation, the child did not follow up at any medical center, and the parents clarified that the child had a normal life with no reported manifestations of medical concern. Both parents are consanguineous with no medical history of concern. She had no history of recurrent infection, no history of blood transfusions, hypertension, diabetes, or other conditions of medical concerns, no history of previous surgeries, and no history of drug allergies. Developmental history was compatible with her age and vaccination was up to date. On examination, she looked pale, fatigued, and not dysmorphic; cardiac examination showed a hemic systolic murmur at the apex; there was no organomegaly, no dermatologic manifestations, no signs of skin inflammation or vasculitis; and neurologic examination and other systems review were unremarkable. Subsequently, she was admitted to the hospital for further investigation and supportive treatment. Her initial blood works showed Hb of 3.8 gm/dL, reticulocytopenia, slightly elevated serum bilirubin, and glucose-6-phosphate dehydrogenase (G6PD) deficiency. She had a positive direct antiglobulin test (DAT++) and was given packed red blood cells (PRBCs) with good improvement (Table 1). The initial impression was autoimmune hemolytic anemia with reticulocytopenia, so the child started on trials of intravenous immunoglobulin (IVIG) 1 gm/kg/day for 2 days and prednisolone 2 mg/kg/day for 1 month and tapering over 2 weeks with no apparent response. Bone marrow aspirate and biopsy were performed and showed reduced erythropoiesis that is consistent with PRCA. Parvovirus and Epstein-Barr virus (EBV) DNA by polymerase chain reaction (PCR) were not detected. The patient was kept under close follow-up at the clinic while preparing for the bone marrow transplantation process. She has a human leukocyte antigen (HLA)-matched sibling donor. Meanwhile, whole exome sequencing (WES) was requested aiming at a final solid diagnosis. During that time, the patient required weekly blood transfusion, as the HB level was dropping so fast reaching down to 2 gm/dL in some reports. Sequence analysis using the Blueprint Genetics (BpG) Whole Exome Plus identified CECR1 c.714_738dup, p. (Ala247Glnfs*16) a homozygous variant and a heterozygous missense variant G6PD c.563C>T, p. (Ser188Ph). Following the diagnosis, we aimed to start the patient on anti-tumor necrosis factor agents; however, the parents refused this type of therapy, so we are currently preparing the patient for bone marrow transplantation; she is clinically well, with reduced blood transfusion requirements (every 3-4 weeks).
Laboratory Results of the Two Patients.
Case 2
The second case is a 10-year-old Saudi boy, known to have Hodgkin’s lymphoma (HL), who ended chemoradiotherapy 6 years ago. He presented with fever, decreased activity, and poor oral intake; oxygen saturation was 98% room air, blood pressure was normal (120/80), and there was mild tachycardia (115 beats/min). Both parents are consanguineous with no medical history of concern. He had no history of hypertension, diabetes, or other conditions of medical concerns, no history of previous surgeries, and no history of drug allergies. His developmental history was up to his age, and his vaccination was up to date. On examination, he looked ill, fatigued, and not dysmorphic; the cardiac examination was normal, there was no organomegaly nor lymphadenopathy, ENT examination was unremarkable, the neurologic examination was normal, there were no dermatologic manifestations, and there were no signs of skin inflammation or vasculitis. He was admitted to the hospital for further investigation and supportive treatment. His initial blood works showed Hb of 10 gm/dL, white blood cells 7000, and the absolute neutrophil count (ANC) was 0.5. Liver enzymes and electrolytes were within the normal range. The initial impression was infection-induced bone marrow suppression, and the child was treated as a case of fever in immunocompromised patient using intravenous antibiotics. The episodes of fever and neutropenia recurred more than once, so the patient was further investigated for the cause of neutropenia. Other investigations including peripheral blood film, antinuclear antibody, anti-ds DNA antibodies, immunoglobulin level, and virology screening (HIV, EBV, CMV, hepatitis) were reported as normal (Table1). Furthermore, we requested bone marrow aspirate and biopsy that showed no apparent malignancy nor clonal proliferation. Moreover, a molecular panel study for congenital neutropenia was also reported as normal. WES was requested as a possible diagnostic tool and showed a homozygous ADA2 mutation c.1447_1451del, p. (Ser483Profs*5). The patient is under close follow-up at the clinic while preparing to start anti-tumor necrosis factor (TNF) agents.
Discussion
The whole-exome plus identified two different mutations in our two reported cases. In the first case, CECR1 c.714_738dup, p. (Ala247Glnfs*16) a homozygous frameshift variant and a heterozygous missense variant G6PD c.563C>T, p. (Ser188Phe) were identified, and to the best of our knowledge, this is the first time to report this CECR1 mutation. The second patient was homozygous for ADA2 c.1447_1451del, p. (Ser483Profs*5), and this mutation has been reported only once before, by Alabbas et al 3 Hematological manifestations have been identified in association with DADA2 in several reports. One hundred fifty-three patients were reviewed who are included in 28 publications for patients with deficiency of DADA2, where their presentation included one or more hematological manifestations. One hundred two patients (66.6%, female n=52) presented with hematologic manifestations. The median onset of disease was at 5 years of age, range 1 month to 52 years. It was noticed that different types of anemia including autoimmune hemolytic anemia, Evans syndrome, pure red cell aplasia, anemia, and Diamond-Blackfan anemia like features were the most frequent presentation occurring in 53% of patients, in which PRCA constitutes 12.7%, autoimmune hemolytic anemia 3.9%, Evans syndrome 1.96%, anemia 14.7%, and DBA was 18.6%. This was followed by lymphopenia and organomegaly (splenomegaly ± hepatomegaly), 32% each. Of particular concern, our first patient has PRCA which was the main presentation in 12 patients with atypical features of Diamond-Blackfan anemia. Our second patient presented with neutropenia and previously treated HL knowing that HL has been reported in 2 cases only from the total of 102 patients. Four patients were successful on hematopoietic stem cell transplant (HSCT), 2 on anti-TNF, 2 failed on corticosteroids, and 2 failed on anti-TNF, whereas other patients are maintained on blood transfusion, corticosteroids, or monthly immunoglobulins. Table 2 summarizes DADA2 that presents with hematologic manifestations.
Published Reports and Abstracts of 153 Patients With DADA2 Deficiency and Hematological Manifestations.

Abbreviations: DADA2, deficiency of adenosine deaminase 2; PRCA, pure red cell aplasia; HSM, hepatosplenomegaly; AIHA, autoimmune hemolytic anemia; TNF, tumor necrosis factor; HSCT, hematopoietic stem cell transplant; DBA, Diamond-Blackfan anemia; WGS, whole genome sequencing; ALPS, autoimmune lymphoproliferative disorder; MAS, macrophage activation syndrome; CECR1, cat eye syndrome receptor1; WES, whole exome sequencing; HL, Hodgkin lymphoma; G6PD, glucose-6-phosphate dehydrogenase.
The diagnosis of pure red cell aplasia (PRCA) depends on the presence of severe anemia with peripheral blood reticulocytopenia and absent or reduced erythroblasts in the bone marrow. Our first patient did not have any feature that suggests the congenital DBA form. She presented at age of 5 years which is not usual for patients with DBA, and she did not have any growth retardation or developmental defects. Trials of steroids and immunoglobulin did not improve her anemia; moreover, she had a positive DAT that made it difficult to find compatible blood every time she needed a transfusion.
Subsequently, we preferred to perform more extensive investigations including WES rather than trying other therapeutic options without a solid clear diagnosis. PRCA has been reported a few times in association with DADA2.4,6-8 However, our first case represents a novel ADA2 mutation which is c.714_738dup, p. (Ala247Glnfs*16) a frameshift variant, that duplicates 25 base pairs causing a frameshift, resulting in a premature stop codon at position 16 in a new reading frame in exon 5 (of 10 total exons). Protein truncation or nonsense-mediated mRNA decay are supposed to result in a loss of normal protein function. To our knowledge, this mutation has never been identified or published in the medical literature.
Our second patient presented with persistent neutropenia and was known to have HL who have been treated using chemoradiotherapy and was under follow-up for the past 6 years. HL is a specific type of lymphoma with distinct behavior and clinical characteristics. 31 In classical HL, diagnosis depends on the histologic demonstration of Reed-Sternberg cells that comprise 1% to 2% of the total tumor cell mass with a background of mixed inflammatory cells. 32 Initial investigations of our patient ruled out HL relapse, and the patient was found to have a specific ADA2 mutation. Persistent neutropenia was the only presentation of our patient. Neutropenia has been reported several times in association with ADA2 deficiency.13,15,19 We needed to rule out the relapse as the cause of neutropenia before proceeding with more sophisticated investigations. HL has been reported only once in association with ADA2 deficiency in 2 siblings, 3 and interestingly, ADA2 mutation in these 2 siblings was similar to our patient. Knowing that ADA2 deficiency has been also reported with various lymphoproliferative disorders, this raises a high concern that ADA2 deficiency might have a direct causative effect on developing HL. However, a direct correlation between the two conditions has not been established up until now.
Corticosteroids and possible transfusion support are the mainstays of treatment for congenital PRCA and DBA; however, the only curative modality is stem cell transplant. 2 For acquired PRCA, it is essential to identify patients with myelodysplastic syndrome, drug-induced PRCA, and B19 parvovirus-associated PRCA as these situations necessitate a syndrome-specific therapeutic approach. For other causes of primary and secondary acquired PRCA, the treatment of choice is immunosuppression. Cyclosporine A, with or without steroids, is regarded as the single most effective immunosuppressive agent. 1
On the other hand, the treatment options for DADA2 previously included steroid therapy, which was associated with relapses upon tapering, tocilizumab showed success in some patients; however, its use was associated with recurrent strokes. It was recently believed that anti-TNF agents are an effective treatment. Michniacki et al reported successful treatment with TNF inhibitor in a patient with DADA2 associated with bone marrow failure. 11 Also, successful use of etanercept, immunoglobulin, and low-dose steroids with complete resolution of cytopenia was reported by Ekinci et al. 12 However, it was concluded in one study that complete loss of function in ADA2 seen in hematologic disease is not in favor of anti-TNF treatment, whereas there is a promising response to TNF inhibition in case of vasculitis due to a subtotal loss of function. 13 Thalidomide has been used with better results but was complicated with neurologic toxicities. 33 Hematopoietic stem cell transplant is the definitive treatment, especially when reversal of cytopenia and immunodeficiency are desired. The first successful HSCT was reported by Van Montfrans et al in 2014. 6 Hashem et al reported a successful HSCT treatment of PRCA associated with DADA2 after failing multiple lines of treatment. 4 In addition, in a multicenter study, it was noted that all patients who had HSCT were cured and doing well at an 18-month follow-up. 34
Conclusion
We report two different DADA2 variants in two children; the first presented with PRCA and the second presented with persistent neutropenia. DADA2 is extending phenotypically beyond its known manifestations to include various hematologic presentations. We emphasize the utilization of genetic testing that can lead to a better understanding of different hematologic disorders with improved detection and diagnosis, which will ultimately offer different therapeutic options and prevent unnecessary interventions.
Footnotes
Authors’ Note
The article was presented in abstract form at the annual hematology transfuse cell therapy meeting held in Turkey, October 2020.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval to report this case was obtained from King Salman Armed Forces Hospital in Tabuk (KSAFH-REC-2021-402).
Informed Consent
Parents of both children gave informed verbal consent and agreed to participate in the study; names of the patients and any identifier (photos, radiological images) were not included in the study.