
Abstract
The intellectual disability syndrome characterized by seizures and dysmorphic features was initially described in 2017 and was associated with genetic variants in the OTUD6B gene, identified by exome sequencing (ES) in a large cohort. This multisystem disorder primarily affects the central nervous system, the gastrointestinal, and the skeletal systems. In this article, we describe the first Mexican patient diagnosed by ES. The homozygous c.433C>T (p.Arg145*) variant of the OTUD6B gene confirmed this intellectual disability syndrome. In addition to seizures and other more frequently reported manifestations of this condition, this is the third patient with associated hypothyroidism and hypogammaglobulinemia, underscoring the value of screening for these conditions in other patients. The current challenge with this patient is to ensure medical management of his seizures and provide him with a better quality of life. The possibilities of additional therapeutic approaches may increase by understanding the physiopathology of the involved pathways.
Introduction
The intellectual developmental disorder characterized by a dysmorphic facies, seizures, and distal limb anomalies (OMIM 617452), also known as OTUD6B-related disorder, was initially described in 2017 by Santiago-Sim et al, who associated the role of genetic variants in the OTUD6B gene, 1 identified by exome sequencing (ES) in a large cohort. Later, 3 other cases were detected after a genetic research odyssey was undertaken, also by ES.2 -4
This syndrome is a serious multisystemic disorder characterized by poor general growth, developmental delay, early-onset seizures, intellectual disability, as well as dysmorphic craniofacial and distal extremity features. The phenotypic manifestations are variable, whereby the most severe include a neurodevelopmental disorder with microcephaly, the lack of speech, and an inability to walk, as well as the need for feeding probes. Other manifestations include congenital heart disease or nonspecific abnormalities of the brain. In some cases, intellectual disability is mild to moderate with normal speech and motor development. 1
According to the reported cases, an autosomal recessive inheritance pattern has been established. 1 The OTUD6B (ovarian tumor domain-containing 6B) gene is located on chromosome 8q21.3 and encodes a deubiquitinating enzyme (DUB). 5 DUBs can recycle ubiquitin as components of the 26S proteasome and recover proteins from the degradation pathway by deubiquitination. In addition, this enzyme regulates central biological processes, including DNA repair, apoptosis, oncogene expression and function, and checkpoint regulation. 6 This gene is expressed in the brain, lungs, liver, gastrointestinal tract, skin, testicles, cardiovascular system, adipose tissue, adrenal glands, thyroid, pancreas, and lymphocytes. 7
The OTUD6B gene regulates protein synthesis in cells by operating downstream from the mammalian target of rapamycin complex 1 (mTORC1). 8 The mTOR has been shown to integrate intracellular signals to control cell growth, nutrient metabolism, and protein translation. This pathway regulates neural stem cell differentiation, neural progenitor migration, dendrite development, and neuron maturation. 9
Currently, only 7 different genetic variants have been described (ie. nonsense, missense, and intronic site variants) in 15 cases (9 families) affected by this syndrome, of which the truncating c.433C>T variant (rs368313959) has been identified as the most common (12 of 30 alleles).1 -3 After random mating calculation, the prevalence of this variant in the homozygous state in a Latino population is estimated to be 1/145 000. 1
In this article, we herein report the first Mexican case of OTUD6B-related disorder. The clinical history is provided, and a comparison is made with other cases in the literature.
Case Report
The index case is a 10-year-old Mexican male with unrelated healthy parents. He is the first and only son. The parents came to the clinic searching for treatment of their son’s refractory epilepsy and insomnia. The patient was born by cesarean section at 38 weeks gestation. During the pregnancy, there was no intrauterine growth restriction; at birth, the patient’s weight was 3 200 g and his length was 52 cm, with an APGAR score of 9-9. Four hours after birth, the patient developed perioral cyanosis and was transferred to the intensive care unit with a probable diagnosis of neonatal sepsis. During his hospital stay, an echocardiogram reported patent ductus arteriosus that closed spontaneously. The newborn screening was reported as normal. He was discharged after 11 days.
Since the first days of life, the patient presented recurrent pulmonary infections associated with multiple hospital admissions, severe gastroesophageal reflux, and breast milk intolerance. At 6 months, he underwent a Nissen fundoplication and gastrostomy.
From the age of 7 months until now, the patient has presented atonic seizures and childhood spasms that are difficult to control. He initially responded well to valproic acid, but developed Fanconi syndrome as a secondary adverse event. He also had a good response to oxcarbazepine but developed severe hyponatremia, so both medications had to be withdrawn. Other prescribed medications included phenobarbital, phenytoin, atomoxetine, aripiprazole, quetiapine fumarate, haloperidol, sertraline, pregabalin, olanzapine, topiramate, levetiracetam, mirtazapine, and ethyl loflazepate, but these medications could not control the seizures either or led to adverse reactions such as irritability, insomnia, tremors, and hallucinations, among others. He is currently treated with lacosamide, clobazam, brivaracetam, and acetazolamide, which have decreased the number of seizures (5 events/month). He recently developed hypokalemia secondary to the use of acetazolamide but was successfully treated with potassium supplementation.
Since his first year of life, his weight, height, and head circumference have been below the third percentile. In addition, at 18 months of age, the patient was diagnosed with subclinical hypothyroidism and selective immunodeficiency of immunoglobulin (Ig)G and IgA, as well as nephrocalcinosis.
The primary dysmorphic features included microcephaly, elongated eyelid fissures with eversion of the outer third of the lower eyelid, arched and broad eyebrows, a depressed nasal tip with a short columella, small and spaced teeth, and micrognathia. He had large cup-shaped ears with low implantation, and bilateral retroauricular pits. Polydactyly of the right hand and left foot were present from birth and were treated surgically. He also had bilateral palmar aberrant folds (dermatoglyphics) and a sacral dimple. This clinical picture fulfilled the findings of a suspected diagnosis of Kabuki syndrome (OMIM 147920); however, sequencing the KMT2D gene—the most commonly associated—ruled out the diagnosis.
Since this diagnostic possibility was excluded, ES was performed. It was only carried out in the patient’s amplifying exome (>98% of the coding regions); mitochondrial DNA was sequenced with an Illumina platform. ES resulted in a homozygous variant in the OTUD6B gene, c.433C>T (p.Arg145*). This nonsense variation (rs368313959) has been described in Clinvar and Baylor Genetics as pathogenic, and other databases have reported an allelic frequency of 0.00014 (Genome Aggregation Database) and 0.00015 (Exome Sequencing Project). No other relevant findings were identified. According to the ACMG scoring guidelines, 10 it appears to be a rare variant fulfilling PVS1, PM2, and PP5 criteria, which could be classified as pathogenic.
Currently, the patient weighs 24 kg (below p3) and measures 1.24 m (below p3). He has presented delayed overall development in terms of head support, sitting with support, and with no gait development, he only emits sounds, and obeys simple commands. He is receiving solid foods orally and liquids by gastrostomy. Furthermore, he is well controlled with levothyroxine and gamma globulin for the management of subclinical hypothyroidism and selective immunodeficiency, respectively. Recently, the patient was diagnosed with insulin resistance and allergic colitis. Figure 1 shows the patient’s current facial and hand features.
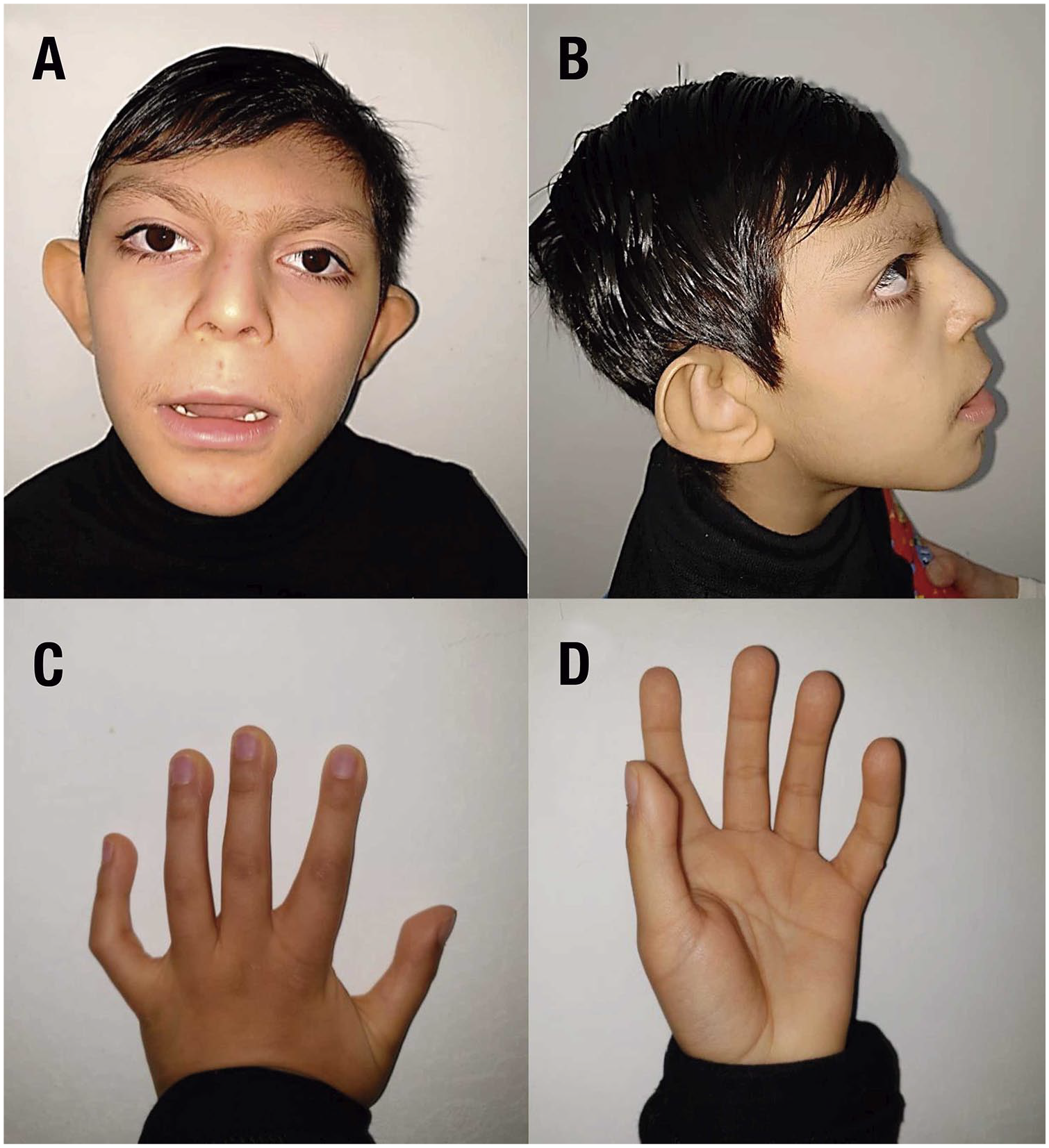
Clinical features of the patient. Photographs illustrating the phenotype of the patient. (A) Front face; (B) lateral face; (C) back of the left hand back; (D) palm of the left hand.
Discussion
We report the case of a Mexican child with severe intellectual disability, seizures, and dysmorphic features, with a molecular diagnosis obtained by ES of a homozygous pathogenic variant in the OTUD6B gene, c.433C>T (p.Arg145*).
Due to the rarity of this syndrome and the scarce literature available, Table 1 presents the clinical and molecular findings of the 16 patients described to date,1 -4 including the present case. This aids in visualizing the similarities and differences among patients in terms of the variants reported previously. In general, the central nervous system, gastrointestinal, and skeletal systems are the most compromised when genetic variants are present in OTUD6B. Moreover, craniofacial dysmorphism, cardiologic malformations, and recurrent respiratory infections are frequently associated clinical features and appear to be dependent on a specific variant (ie, c.433C>T).1,3
Comparative Table of Clinical and Molecular Characteristics Present in Patients With Pathogenic Variants Within OTUD6B.
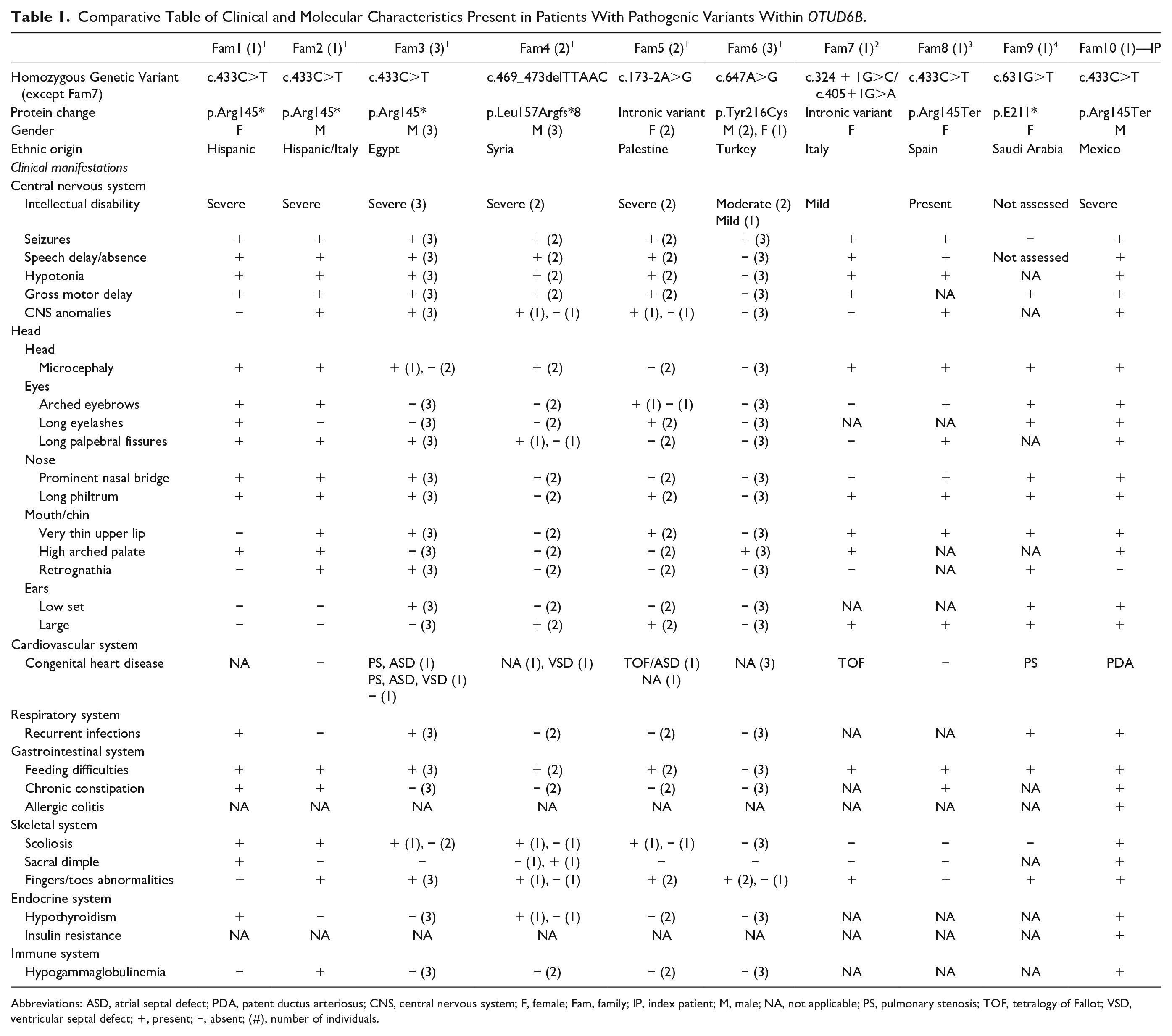
Abbreviations: ASD, atrial septal defect; PDA, patent ductus arteriosus; CNS, central nervous system; F, female; Fam, family; IP, index patient; M, male; NA, not applicable; PS, pulmonary stenosis; TOF, tetralogy of Fallot; VSD, ventricular septal defect; +, present; −, absent; (#), number of individuals.
It is important to highlight that hypothyroidism and IgG and IgA immunodeficiency have been reported previously in other cases. 1 Our patient is the third case with hypothyroidism and the second with hypogammaglobulinemia (Table 1), underscoring the value of screening for these conditions. These findings could be explained by the fact that the OTUD6B gene is expressed in the thyroid and lymphocytes. 7 Also, additional clinical manifestations that had not been previously reported were present in our proband, including allergic colitis and insulin resistance. Further studies must be conducted to confirm their relation with OTUD6B variants.
Our patient’s clinical features were initially compatible with the diagnosis of Kabuki syndrome (OMIM 147920) and consistent with other reports.2,3 Other diagnoses, such as Rubinstein-Taybi syndrome (OMIM 180849) and DiGeorge syndrome (OMIM 188400), among others, had been suggested as differential diagnoses. Consequently, diagnosing cases with partially overlapping complex phenotypes warrants the use of broad genetic tests, such as exome sequencing.
Given the homozygous pathogenic variant identified in our case, the clinical manifestations were consistent with those of 6 other patients (Table 1; Fam 1, 2, 3, 8).1,3 The presence of craniofacial dysmorphism (ie, microcephaly in 5/7 patients, long palpebral fissures in 7/7 patients, and nose features in 7/7 patients), as well as recurrent respiratory infections (5/6 patients; 1 patient not reported), and chronic constipation (4/7 patients) are all clinical features that might be predominantly associated with this variant. Differences between these patients included the presence of congenital heart disease (3/6 patients), as well as the diagnosis of hypothyroidism (2/6 patients) and hypogammaglobulinemia (2/6 patients).
The broad clinical picture is caused by the genetic variant type. For example, variants that lead to the absence of protein are associated with more severe phenotypes, compared with protein conformation changes only (Table 1). The research literature has mainly reported homozygous cases, making it difficult to approach this differential diagnosis of compound heterozygous states given the lack of knowledge on its impact on loss of function, and to the expression of different allele combinations.
Knowing the clinical impact of genetic variants on the organs and systems that could be affected guides the physician to intentionally review them. Therefore, an action plan can be established for the patient’s medical management and improvement in his quality of life.
Determining the most appropriate therapeutic approach in patients with this intellectual disability syndrome has represented a challenge in terms of patient management, particularly in seizure control.
Furthermore, gastrointestinal and cardiologic manifestations, as well as motor and speech delays, can benefit from early treatment to ensure a better quality of life. Multiple standard treatments have been used but are associated with the development of adverse effects.
The presence of the c.433C>T variant in the homozygous state seems to severely affect the structure and function of the central nervous system.1,3 This could explain why our patient had difficult to control seizures, severe mental retardation, and an abnormal corpus callosum. OTUD6B regulates protein synthesis in non–small cell lung cancer cells, operating downstream from mTORC1. In the past few years, an increase in preclinical data has uncovered the molecular pathway of the mTOR, which appears to be crucial in many genetic and acquired epilepsy syndromes; it has been shown to integrate the intracellular signals required to control cell growth, nutrient metabolism, and protein translation.9,11 The mTOR complexes are essential in neurogenesis and in the establishment of neural circuits. This pathway regulates neural stem cell differentiation, neural progenitor migration, dendrite development, and neuron maturation. 9
Different animal models in which mTOR was hyperactivated showed that the inhibition of mTORC1 with rapamycin and other compounds, such as vigabatrin, valproic acid, and a ketogenic diet, resulted in the prevention, delay, and decrease in the number of seizures.11,12 Future studies must be conducted to evaluate whether the intervention of this pathway in patients with OTUD6B could further support the available therapeutic options.
Conclusion
We report the first Mexican case of the OTUD6B-related disorder, as well as the seventh patient (fourth family) worldwide, with the homozygous variant c.433C>T (p.Arg145*). The current challenge in these patients is their medical management. Our patient also had hypothyroidism and immunodeficiency, findings consistent with other reported cases. Moreover, allergic colitis and insulin resistance could be new expressions of the syndrome that need to be evaluated in future cases. Given our initial clinical diagnosis of Kabuki syndrome, due to the presence of craniofacial dysmorphism, limb abnormalities, and intellectual disability, and considering other authors’ initial clinical diagnoses (eg. Rubinstein-Taybi syndrome), we propose a genetic panel that includes the 3 syndromes, to shorten the diagnostic odyssey. Even if ES is the indicated approach in complex phenotypes, the interpretation of genetic variants using a broad test such as this may lead to difficulties in medical practice. Thus, a specific, targeted genetic test would be a preferable choice.
Footnotes
Acknowledgements
We would like to thank Dr Olaf Bodamer for his medical advice in this case.
Authors’ Note
Specific data are available on request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval when reporting individual cases.
Informed Consent
Written informed consent was obtained from parents for patient information to be published in this article. Parents provided permission and signed a consent form permitting full-face sharing in this article.