
Abstract
Dysphagia can be one of the manifestations of inflammatory myopathies (IMs). In some patients, it can be one of the presenting symptoms or the only symptom. We present a patient with dysphagia and progressive muscle weakness who was eventually diagnosed with inclusion body myositis (IBM). Treatment with oral steroid provided no major improvement in symptoms and thus was eventually stopped. Dysphagia in IMs is associated with complications and poor prognosis. A multidisciplinary approach is needed in its diagnosis and management as this report exemplifies.
Introduction
Inflammatory myopathies (IMs) are a group of rare immune-mediated diseases characterized by muscle weakness. They include dermatomyositis (DM), polymyositis (PM), and necrotizing myopathy.1-3 Although unlike DM and PM, inclusion body myositis (IBM) is non-responsive to immunomodulatory therapy. In Caucasians with age above 50, the most common acquired muscle disease is IBM. 4 Dysphagia has been reported to occur in 10% to 73% of these patients and can present at any time during the disease process, sometimes preceding weakness of extremities.2,3 Specifically in IBM, the overall incidence of dysphagia has been reported in literature to be as low as 40% and as high as 80%. 4 Dysphagia in IM primarily affects the skeletal muscle–activated oropharyngeal phase of swallowing. This occurs due to weakness of oropharyngeal, laryngeal, and cervical esophageal musculature. 3 Recognizing this specific type of dysphagia, referred to as “transfer dysphagia,” is important as it is associated with nutritional deficits, aspiration pneumonia, decreased quality of life, and poor prognosis.2,5,6 Patients with IM and dysphagia are reported to have a 1-year mortality rate of 31%. 5 We report a case of a 63-year-old Hispanic female who initially presented with dysphagia and progressive weakness who was eventually diagnosed with IBM.
Case Presentation
A 63-year-old Hispanic female with past medical history of osteoarthritis and gastroesophageal reflux disease presented to the gastroenterology clinic for dysphagia for the past several months. The patient has been complaining of a progressively worsening of the dysphagia occurring at the time of initiating a swallow. She has also noted unintentional, non-quantifiable weight loss. She denies any abdominal pain, diarrhea, constipation, hematemesis, melena, and hematochezia. On further questioning, she mentioned that she was having progressive weakness for 5 years accompanied by proximal muscle weakness and recurrent falls. She denied smoking, alcohol, or illicit drug use. Family history is unrevealing. Given the diffuse and evolving nature of the accompanying muscle weakness, neurology was consulted.
On general and neurological examinations, vital signs were within normal limits. She has soft non-tender abdomen. She had mild ptosis on examination. Weakness was noted on shoulder abduction (4+/5), elbow extension and flexion (4/5), finger flexion (left worse than right), and hip flexion weakness (4/5), with difficulty rising from a seated position. Sensory examination and deep tendon reflexes were normal. Skin examination was normal, including no heliotrope erythema, Gottron’s papules, or periungual telangiectasia.
On laboratory examinations, Creatine Kinase (CK) (613 IU/mL) and aldolase (16.5 U/L) levels were elevated. Complete blood count and comprehensive metabolic panel were within normal limits. Thyroid-stimulating hormone, cortisol level, erythrocyte sedimentation rate (ESR), and C-reactive protein (CRP) were unremarkable. Acetylcholine receptor antibody, muscle-specific kinase antibody, myositis panel, antinuclear antibodies (ANA), RNP, SSA IgG, SSB IgG, and smooth muscle antibodies were negative. Nerve conduction study and electromyography did not indicate a myopathic process.
A modified barium swallow showed a narrowing at the cricopharyngeal (CP) area (Figure 1). An upper GI (gastrointestinal) endoscopy was done which showed LA Grade D esophagitis and a 4 cm hiatal hernia. Although there was no specific stenosis of the upper sphincter area by esophagogastoduodenoscopy (EGD), we decided to be aggressive and a dilation was performed through the scope balloon dilator to a diameter of 20 mm at the level of the cricopharyngeus, which resulted in only a minimal improvement in symptoms (Figure 2).
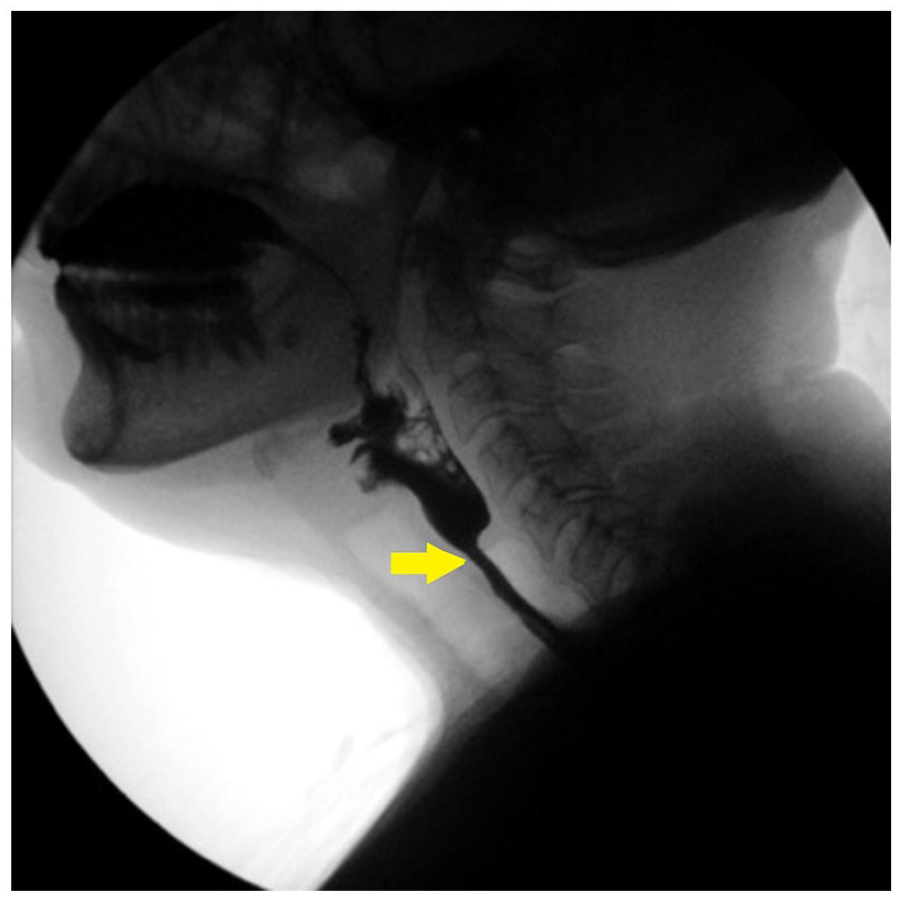
Modified barium swallow showed a narrowing at the cricopharyngeal area (as indicated by the yellow arrow on the image).

Esophagogastroduodenoscopy image post-dilation showing mild bleeding and mucosal injury (indicated by yellow arrows), at the level of the upper esophageal sphincter. Mild bleeding and mild mucosal injury are expected post-dilation.
A subsequent left biceps brachii biopsy was done. The skeletal muscle demonstrated marked endomysial inflammation by mononuclear inflammatory cells (mostly of lymphocytes and occasional histiocytes). Some muscle splitting and scattered muscle atrophy, necrosis, and regeneration were also identified (Figures 3 and 4). No rimmed vacuoles, ringed fibers, or inclusion bodies were identified. Rare congophilic inclusions are seen. A Gomori trichrome (GT) stain was performed to reveal scattered ragged-red fibers. A succinate dehydrogenase stain reveals scattered ragged-blue fibers, corresponding to the previously detected ragged-red fiber in the GT stain. Cytochrome c oxidase was noted to be negative in scattered fibers. However, a lack of rimmed vacuoles and exceeding rare congophilic inclusions limited a histologic diagnosis of IBM. A major histocompatibility complex (MHC) class I antigen immunohistochemical stain and electron microscopy were not performed.

This skeletal muscle (left bicep) shows infiltration by mononuclear inflammatory cells. Scattered muscle necrosis, as noted in between the yellow arrows, is identified by pale, pink muscle fibers. Muscle fibers vary in diameters (5-100 µm), compatible with scattered muscle atrophy. Finally, patchy expansion of the endomysium is present due to the inflammatory infiltrates (4× magnification).

This skeletal muscle (left bicep) shows infiltration by mononuclear inflammatory cells, consisting mostly of lymphocytes and occasional histiocytes (indicated by yellow arrow). Muscle fiber necrosis is noted by a pink, pale appearance, compared with surrounding fiber. No rimmed vacuoles, ringed fibers, or inclusion bodies are identified (40× magnification).
The final pathology diagnosis was an IM with denervation atrophy associated with re-innervation and type 2 muscle fiber atrophy. Based on the non-conclusive biopsy results for IBM, patient was started on oral prednisone 40 mg daily.
On follow-up visits, no major improvement has been noted in both her dysphagia and weakness. Clinically defined IBM diagnosis was given based on the European Neuromuscular Center (ENMC) IBM research diagnostic criteria 2011. Prednisone was slowly tapered and discontinued.
Discussion
In the GI tract, the oropharynx and the upper esophagus (striated muscle portion) are most commonly involved in IMs. Transfer dysphagia is the most common symptom, with a wide range of prevalence of 10% to 73% in known literature.2,5,7-9 In patients with IBM, the overall incidence of dysphagia ranges from a low of 40% to a high of 80%. 4 Dysphagia can be the initial presenting symptom in IMs, and in some cases, it can be the only manifestation. In the absence of facial, bulbar, or proximal muscles, this can lead to delays in diagnosis.2,8 Hence, clinicians should consider IMs in cases of patients with unexplained “transfer type” dysphagia. In a study of 783 patients with IM, 62 patients were found to have dysphagia. In this study, 96% reported difficulty with solid and dry food, 85% had globus sensation, and 75% had coughing while eating with concomitant risk for aspiration. Involvement of the striated muscle of the hypopharynx and upper esophageal sphincter is the main cause of “transfer” dysphagia in IMs. The underlying pathophysiologic mechanisms include edema, chronic inflammation, atrophy of esophageal muscles, and at rare times, spontaneous esophageal rupture. 8 The prompt recognition of dysphagia in patients with IM is important as delays in diagnosis can be associated with various complications such as nutritional deficits, aspiration, decreased quality of life, and poor overall prognosis.2,5 The nutritional challenges may lead to placing a percutaneous gastrostomy tube to avoid aspiration, maintain nutrition, and provide access to give medications.
Serum CK elevation is seen in approximately 75% of patients with IM complaining of dysphagia and has a high positive predictive value. Given its high positive predictive value, CK levels can be obtained as a screening test in any new case of pharyngeal dysphagia of uncertain etiology; ESR and ANA can also be elevated. 5 Myositis-specific antibodies are less sensitive, but have high specificity and can be helpful in establishing the underlying diagnosis. 10 To evaluate swallowing, modified barium swallow with videofluoroscopy is the diagnostic test of choice. 11 Restrictive CP abnormalities, either CP bar, stenosis, or Zenker’s diverticulum, were also evident radiographically in 69% of cases with IM. 5 High-resolution manometry may show abnormally low esophageal sphincter pressures with normal relaxation and low-amplitude non-peristaltic contractions.8,12 However, in a study of 53 adult patients with IM, it was found that manometric involvement is poorly correlated with the esophageal symptoms. 13 Upper endoscopy findings may include esophagitis and strictures, and are regarded as non-specific. 12 Muscle biopsy is considered the gold standard for diagnosing myositis. 5 A multidisciplinary approach is necessary to differentiate the muscular dysphagia associated with IM from dysphagia caused by acute cerebrovascular accident and other neuromuscular disorders. 8
Initial management strategies initially instituted for patients with this type of “transfer” dysphagia include diet modification, speech pathologist evaluation and modification of the consistency and quantity of food and fluid in the diet, and coaching on positioning techniques to improve swallowing. 8 However, these interventions lack definitive studies with regard to their effectiveness in IM. 2 There is a lack of randomized control trials that evaluate the efficacy of glucocorticoids or immunosuppressive agents in IM patients presenting with dysphagia. Despite evidence being largely anecdotal, glucocorticoids remain a mainstay of treatment. 8 In addition to glucocorticoids, immunosuppressive agents such as methotrexate, cyclosporine, azathioprine, and cyclophosphamide can also be utilized.8,9 In cases of steroid-resistant dysphagia in PM and DM patients, intravenous immunoglobulin (IVIG) has been used successfully in the management of life-threatening esophageal involvement.8,14,15 Evidence is lacking with regard to the effectiveness of glucocorticoids and immunosuppressive agents in treating dysphagia in IBM patients.2,8 The use of IVIG in IBM has been reported to coincide with some improvement in dysphagia. Although the studies investigated IVIG in patients with moderate or severe IBM and the benefit reported was short lived, 4 CP myotomy appears to be beneficial in patients with IBM.2,8 One concern in our patient was the presence of gastroesophageal reflux disease (GERD) and esophagitis where a CP myotomy could result in the possibility of aspiration of refluxed contents particularly nocturnal gastroesophageal reflux. Other interventions that have been tried such as balloon dilation and botulinum toxins have variable success.2,8 Alternative route for nutrition is considered if the above-mentioned measures fail. Enteral nutrition can be achieved via nasogastric or nasoenteric tube, percutaneous endoscopic gastrostomy (PEG), or jejunostomy tubes. 8 In 1 study, however, it was found that patients who required PEG tubes have higher mortality (64%). 2
Given its complexity, diagnosing and treating dysphagia in the setting of IMs require a multidisciplinary approach, including a gastroenterologist, neurologist, rheumatologist, otolaryngology surgeon, nutritionist, and rehabilitation medicine doctor. Our experience with this patient emphasizes these management principles.
Footnotes
Author Contributions
M.E. wrote the case report and the discussion part for the manuscript. D.K. provided inputs especially on the neurological aspects of the case. O.P. provided images and helped with the pathology discussions. R.M. helped in refining the case and providing expert opinion in the manuscript.
Article Guarantor
Richard McCallum, MD, is the guarantor for the contents of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
Disclaimer
This manuscript is original. It has not been published before nor is any part of it under consideration for publication at any other journal.