
Abstract
Leukocytoclastic vasculitis (LCV) is a rare vascular inflammatory condition that affects post-capillary venules. Its incidence in the pediatric population is unknown. However, its incidence has been shown to increase with age. The causes of LCV can be varied, ranging from drugs to infections to systemic disease. LCV as a presenting symptom of inflammatory bowel disease (IBD) is rare, especially in the pediatric population. A 15-year-old female with a family history of systemic lupus erythematosus was transferred to our hospital with a month-long history of rash, joint swelling and tenderness, periorbital edema, weight loss, and diarrhea. She presented with the objective findings of a biopsy showing LCV and a computed tomography scan read that was concerning for IBD versus infectious colitis. She had a thorough workup, involving both the rheumatology and gastroenterology services, and was ultimately found to have Crohn’s disease. This case reveals the importance of recognition of a constellation of symptoms in IBD even when they are not classical in nature at initial presentation.
Keywords
Introduction
Leukocytoclastic vasculitis (LCV) is a rare, cutaneous, small vessel vasculitis that affects post-capillary venules. The rash is caused by the combination of immune complex deposition and the activation of the complement system. 1 Studies on the incidence show that it is more common in the adult population, with increasing incidence with age. 2 While the majority of cases are idiopathic, infections and drugs are known triggers. A smaller subset of cases is seen in individuals with systemic disease, such as inflammatory disorders including inflammatory bowel diseases (IBD). Classically, it manifests as a palpable purpuric rash that begins on the lower extremities.
In the pediatric population, IgA (immunoglobulin A) vasculitis, also known as Henoch-Schönlein purpura, is the most common form of LCV. The current literature reveals a paucity of information regarding the association of LCV with other systemic diseases in children. We present a case of a 15-year-old female whose first symptom of Crohn’s disease was a palpable purpuric rash that began on her lower extremities.
History and Physical
An obese 15-year-old female with a history of mild intermittent asthma and a family history notable only for 2 first cousins with systemic lupus erythematous was admitted to a general pediatric hospitalist service after a month-long illness course of unknown origin. She originally developed a nonpruritic, painless rash on her feet and hands with bilateral knee pain and ankle swelling. Her pediatrician prescribed a dose of bactrim and a 5-day course of prednisone, which slightly improved her symptoms. However, 2 weeks later the rash worsened, and she was prescribed a course of clindamycin. Within this time period, her rash had ascended to include her shins and thighs, and she had developed bilateral hand edema with new-onset vomiting and non-bloody diarrhea. She presented to an outside hospital emergency department where she was given a course of prednisone 40 mg for 5 days, doxycycline, and nitrofurantoin. Her diagnosis was not determined at any of these previous visits, and despite multiple antibiotics she was not improving. She was referred to dermatology, where a punch biopsy of the rash was obtained. The biopsy was microscopically analyzed at an outside dermatopathology clinic and revealed areas of red blood cell extravasation, vessel wall necrosis, and neutrophil rich infiltrate. However, before the biopsy results of her LCV were finalized, she again presented to an outside emergency department, this time with bilateral periorbital edema, body aches, and severe diffuse abdominal pain with intractable vomiting. An abdominal computed tomography (CT) was obtained, which showed mucosal enhancement with mucosal edema and wall thickening of the right colon, hypervascularity in the mesenteric side of small bowel loops, and wall thickening of the distal sigmoid and rectum, all suggestive of inflammatory enterocolitis.
Given these findings, she was prescribed a course of cephalexin and ondansetron. Throughout her illness progression she remained afebrile, had not had any recent exposures to infectious causes, and no close contacts were experiencing any similar symptoms. By the time she was admitted to our hospital she had lost 20 pounds of weight due to her emesis and decreased appetite.
At presentation she weighed 104.7Kg (CDC 99.27th%tile) and was 170cm tall (CDC 88th%tile) and her vital signs were notable for hypertension with BP 167/98mmHg (95th%tile for age and height is 127/82mmHg), HR 110bpm, T 37.1°C, and RR 20bpm. She was alert, but tired, with appropriate responses on examination. Thrush was noted on her mouth and lateral tongue; however, no mouth ulcers were visible. Her abdomen was diffusely tender to palpation with voluntary guarding; however, no organomegaly was palpated. There was no rebound tenderness, Murphy’s sign, or Rovsing’s sign. She elicited tenderness to palpation over all finger joints, and at her left great toe. Only her right hand was diffusely edematous, without overlying erythema or warmth. She had palpable erythematous purpura diffusely scattered circumferentially over her bilateral lower extremities to her superior thighs, and bilateral upper extremities to her superior shoulders, but spared her palms and soles (Figures 1 and 2).
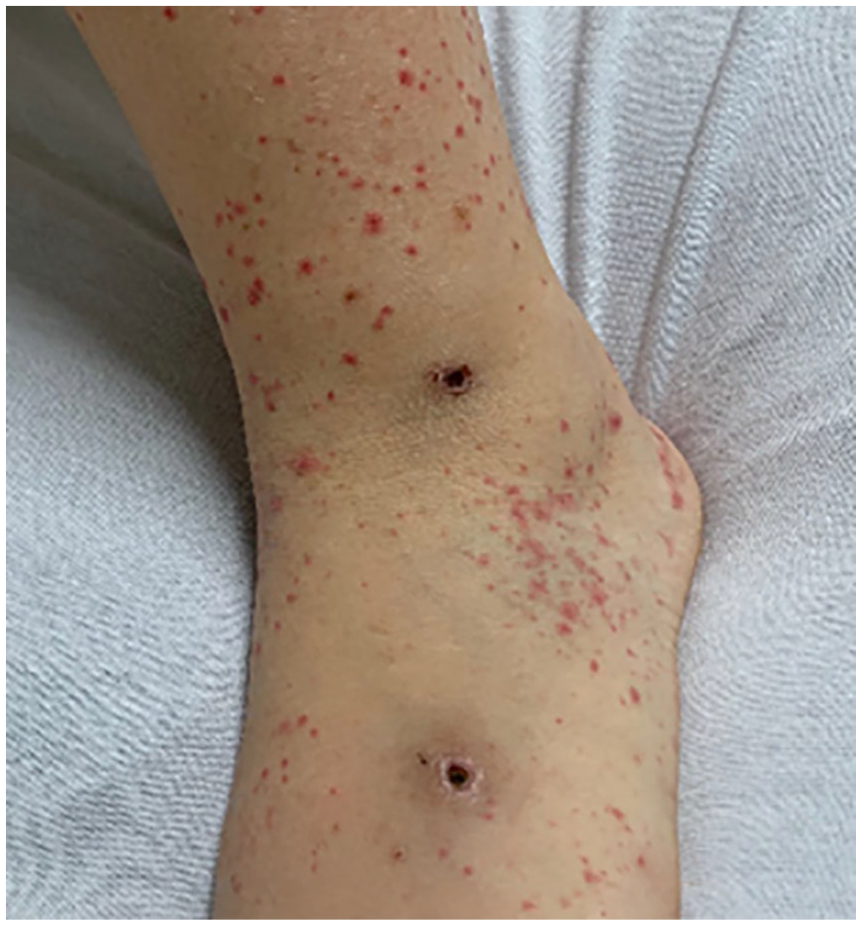
Palpable purpura on left lower extremity with original sites of ulceration.

Palpable purpura on left hand, spared palms.
Diagnostics and Assessment
Her laboratory results showed a leukocytosis (16,600/µL (ref 4.5-13.5)), hypoalbuminemia (2.7 g/dL (ref 3.5-4.9)), and microcytic anemia (11.3 g/dL (ref 12-16), mean corpuscular volume 77.3 fL (ref 78-102)), with elevated C-reactive protein (5.4 mg/dL (ref <1)), and erythrocyte sedimentation rate (27 mm/h (ref 0-20)). Her urinalysis had many red blood cells, white blood cells, and leukocytes with a urine protein to creatinine ratio of 0.25 mg/mg (ref <0.2). Due to concern for renal involvement in the setting of Henoch-Schönlein Purpura, a retroperitoneal ultrasound was ordered. Her kidneys were normal bilaterally, and her UPC ratio subsequently decreased to 0.19mg/mg within twenty-four hours. A urine culture was sent as a follow up to her urinalysis and revealed 25 000-50 000 Col/mL gram negative rods (culture never speciated), so she was started on a five day course of Bactrim DS. Von Willebrand factor, anti-nuclear antibody (ANA), anti-phospholipid, and anti-cardiolipin antibodies were ordered due to concern for a rheumatologic process; however, they were all within normal limits. Given the outside computed tomography findings, a gastroenteritic process was simultaneously worked up. A fecal occult was positive with a fecal calprotectin of greater than 1250 (ref <50). Clostridium difficile and human immunodeficiency virus (HIV) labs were both negative. With the help of the gastroenterology consultants, an esophagogastroduodenoscopy was completed, which showed an ulceration on the roof of her mouth and patchy, mildly erythematous mucosa without bleeding in the gastric antrum and first portion of the duodenum. A colonoscopy revealed scattered aphthous ulcers in the rectum, descending and transverse colon, all less than 0.5 cm in size. The total Simple Endoscopic Score for Crohn’s Disease (SES-CD aggregate), which takes into account the size of the ulcers, the total ulcerated surface area, as well as the extent of stenosis, was 15, which is on the cusp of moderate to severe endoscopic activity (7-15 indicates moderate endoscopic activity and >15 indicates severe endoscopic activity). She, therefore, was diagnosed with Crohn’s Inflammatory Bowel Disease.
She was started on a course of daily solumedrol and transitioned to an extended prednisone taper, with the long-term treatment plan including daily Adalimumab and omeprazole.
Discussion
Leukocytoclastic vasculitis is rare, with approximately 30 to 45 cases per 1 million people.1,3 It is also known as a hypersensitivity vasculitis, which causes swelling, neutrophilic invasion, and even fibrinoid necrosis in the small vessels in the post-capillary venules. 4 This rash, while rare, can be triggered by drugs, malignancy, infections, or other systemic disease. Most frequently, it is idiopathic in nature. 1
The underlying pathogenesis of LCV and the relationship with IBD is poorly understood. Studies postulate that LCV is a result of the interplay between environmental and genetic factors. An inciting factor such as an infection or circulating immune complex creates the immune system response that results in LCV. Antigen excess results in antibody formation, and the subsequent formation of antibody antigen complexes deposit in the blood vessel walls. A resulting cytokine reaction by chemotactic factors (C3a, C5a) cause neutrophilic migration, release of adhesion molecules and cytokines, as well as oxygen-free radicals that lead to vessel wall destruction. 5 Patients who are antineutrophil cytoplasmic antibodies (ANCA) positive have immune complex-free vessel walls on biopsy. This indicates that ANCA antibodies may also be able to trigger the cascade that results in vasculitis and vessel wall histopathology without immune complex involvement. 6
The differential for LCV is broad and workup should be thorough. If there is concern for systemic involvement doctors should obtain a complete blood count, comprehensive metabolic panel for renal and liver involvement, blood cultures, urinalysis to evaluate for Henoch-Schönlein purpura, as well as hepatitis testing, HIV screening, streptococcal infection testing, chest X-ray to evaluate for lymphoma, anti-nuclear antibody, rheumatoid factor, cryoglobulins, serum protein electrophoresis, and serum complements.1,5 If there is low suspicion for systemic involvement, a complete blood count, comprehensive metabolic panel, and urinalysis is a good screen in addition to obtaining a skin biopsy to confirm the diagnosis of idiopathic LCV. Systemic vasculitis should be suspected with fever, night sweats, weight loss, chills, shortness of breath, cough, abdominal pain, bloody diarrhea, or neurologic symptoms. 5
Most patients have spontaneous resolution of their LCV; however, approximately 10% progress to have chronic disease. Supportive care has been shown to be useful in the management of LCV including antihistamines and rest, ice, compression, and elevation. Some require steroids of prednisone or methylprednisolone 1 mg/kg/day with a taper over 4 to 6 weeks. Some may require immunosuppressive or biologic therapy as well.1,3
This case had an interesting presentation that explores the differential diagnosis for a LCV rash in the setting of a family history of rheumatologic problems, and a patient who presents with joint pain, diarrhea, and significant acute weight loss. The initial differential diagnosis included idiopathic LCV, drug-induced rash, rheumatologic disease, IBD, HIV, Clostridium difficile, and Henoch-Schönlein purpura. This differential diagnosis was narrowed as a result of various laboratory findings including microcytic anemia, hypoalbuminemia, leukocytosis, and an elevated erythrocyte sedimentation rate and C-reactive protein. Her CT abdomen showed concern for inflammatory enterocolitis. The Rheumatology and Gastroenterology teams were consulted, and both had high suspicion for IBD based on her laboratory findings and imaging. An upper and lower endoscopic evaluation concluded their diagnosis of Crohn’s disease. Interestingly, her ascending colon and terminal ileum showed the most radiological involvement, but the colonoscopy showed equivalently affected areas from the terminal ileum to the rectum.
Conclusion
Leukocytoclastic vasculitis has a broad differential and is typically idiopathic in nature. However, in this case a teenage girl with a family history of autoimmune disease and new LCV was found to have Crohn’s disease. This diagnosis was made in the setting of microcytic anemia, hypoalbuminemia, elevated inflammatory markers, and clinical findings of joint pain, abdominal pain, and diarrhea. This case presentation reveals the importance of recognition of a constellation of symptoms in IBD even when they are not classical in nature at initial presentation.
Footnotes
Author Contributions
Drs Ford, Mooney, and Shah conceptualized the report, gathered information, drafted the initial manuscript, and reviewed and revised the manuscript. Dr Jenkins coordinated and supervised the information acquisition, assisted with analysis, and critically reviewed and revised the manuscript. All authors approved the final manuscript as submitted and agreed to be accountable for all aspects of the work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.