
Abstract
Cardiac disease is a well-established manifestation of myotonic dystrophy type 1 (DM1), characterized by progressive cardiac conduction slowing with increased risk of atrial and ventricular arrhythmias, heart block, and sudden cardiac death. Multiple disease modifying therapies are in clinical trials for DM1 and show promise in improving skeletal muscle weakness and myotonia. Testing the effects of these medicines, or others, in the heart is of critical importance, but requires identification of endpoints of cardiac function that accurately reflect the state of DM1 cardiac disease. To better define cardiac endpoints, the Myotonic Dystrophy Clinical Research Network (DMCRN) and the Myotonic Dystrophy Foundation (MDF) convened a workshop entitled ‘‘Cardiac Endpoint Workshop’’ in May 2025 at the Myotonic Dystrophy Foundation International Conference. Here, we summarize the discussion at the workshop and perform secondary analysis of cardiac outcomes in the published literature to evaluate cardiac endpoints for clinical impact and trial feasibility. This analysis demonstrates that major cardiac events are too infrequent (<1% annual incidence), and alternatives such as composite endpoints or progression of cardiac conduction prolongation would likely be underpowered in a conventional clinical trial. Given these limitations, we identify areas for further natural history study to better describe longitudinal cardiac structural and functional changes to inform specialized patient selection or identify alternative measures with sensitivity to detect therapeutic impact in a trial.
Keywords
Introduction
Myotonic dystrophy type 1 (DM1) is an inherited disorder caused by a CTG repeat expansion in the 3’ untranslated region of the DMPK gene. It is the most common adult-onset muscular dystrophy, with an estimated prevalence of ∼1/5000.1–4 It is characterized by skeletal muscle myotonia, progressive muscle weakness, cardiac arrhythmia, gastrointestinal motility abnormalities, early cataract development, and CNS involvement, leading to early mortality. Cardiac involvement is estimated to be present in 30–75% of individuals with DM1 and is the second leading cause of death after respiratory failure.5–9 Cardiovascular manifestations most commonly include electrical abnormalities, including conduction slowing and atrial or ventricular arrhythmias, but arrhythmogenic or dilated cardiomyopathy can also occur.
Given the predominance of conduction disease, the largest cohort studies have examined cardiac disease burden in DM1 primarily with surface electrocardiograms (ECG), an example being the prospective natural history study by Groh et al., published in 2008. Other cohorts have performed broader evaluations including intracardiac electrophysiologic recordings, echocardiography, or cardiovascular magnetic resonance (CMR). Many of these studies have found evidence of functional impairment (decreased ejection fraction, wall motion abnormalities, etc.) as well as structural changes (fibrosis, edema, etc.) but general conclusions are limited due to relatively small samples sizes or trial design.
There have been mixed reports on the correlation of CTG repeat length with the frequency and severity of cardiac manifestations and risk of sudden cardiac death.5,10–14 In a prospective cohort of 406 individuals with genetically confirmed DM1, patients with severe ECG findings at baseline had on average longer CTG repeats (606 vs. 704 CTG repeats; p = 0.03), but the repeat length did not correlate with risk of sudden or all-cause death during the study period. To the contrary, a recent longitudinal study of 855 individuals with DM1 found that when the total cohort was divided by 500 CTG repeat steps, higher repeats correlated with higher mortality (37% vs. 19–22%), increased pacemaker implantation, and an increased incidence of sudden death. 15
Over the past 2–3 years the therapeutic landscape for DM1 has quickly evolved with nearly 10 active clinical programs and numerous pre-clinical programs. The focus of nearly all the ongoing clinical therapeutic development is to evaluate for effects in skeletal muscle, and no studies have been designed to investigate therapeutic effects in the heart. As these programs reach their conclusion, with several expected to result in novel drug approvals, the scope of therapeutic impact will need to broaden. One of the next logical targets is to modify the risk of sudden cardiac death and cardiac disease in DM1, as this represents one of the primary drivers of early mortality in the disease.8,16
Recognizing the need to further characterize the cardiac phenotype in DM1 and identify cardiac outcomes or biomarkers to assess therapeutic response in the heart, the DMCRN in partnership with the MDF convened a workshop on May 1st, 2025, in Indianapolis, Indiana, entitled “Cardiac Endpoint Workshop.”
Methods
The “Cardiac Endpoint Workshop” consisted of experts in electrophysiology, advanced imaging, and neuromuscular medicine representing multiple institutions from across the world, ensuring a broad clinical and geographic perspective. The discussion points presented here reflect a structured thematic consensus derived from a moderated plenary session. To ensure the independence of the directive framework, representatives from the pharmaceutical and biotech industries participated in the workshop as observers only.
To report on the content of the workshop, we first summarize and editorialize the topics discussed, which were articulated into four main sessions: i. Impact of cardiac dysfunction in DM1, ii. Mechanism of cardiac dysfunction in DM, iii. Measuring cardiac arrhythmias in clinical trials, and iv. The role of cardiac imaging and exercise in clinical trials.
Next, we synthesize the material discussed in the workshop as well as published literature to develop a framework to organize cardiac endpoints by clinical relevance/impact and feasibility to help guide trial design and identify areas where further natural history data may better identify and define endpoints. We performed a secondary analysis of data from landmark DM1 studies in the literature. Our analytical approach included: 1) Summarization of endpoint definitions to create a scaled hierarchy of severity/risk association; (2) Estimation of weighted annual event rates for cardiac endpoints to inform sample size and feasibility; and (3) Power and detectable-effect analyses for longitudinal ECG changes. The severity/risk association was scaled 1–5 by expert judgment, with 5 being the most severe, (i.e., sudden cardiac death). The secondary statistical analysis of the literature was conducted with input from an independent senior statistician to mitigate any institutional or industry-related bias.
Time-to-event endpoint simulation
We evaluated the feasibility of several clinically relevant cardiac endpoints in DM1 using time-to-event analysis. The endpoints evaluated include sudden death, severe ECG abnormalities, pacemaker or ICD implantation, left ventricular (LV) dysfunction, composite cardiac events, and development of ECG interval prolongation (PR >240 ms and QRS >120 ms). Event rates were abstracted from published studies, and annualized rates were calculated by dividing the number of events by total person-years of follow-up. For studies without total person-years, values were estimated from sample size × median follow-up duration. Random-effects meta-analyses of the log-transformed annualized event rates were performed using the restricted maximum likelihood (REML) method to estimate pooled rates and between-study heterogeneity (I2).17,18 Weighted mean, minimum, and maximum rates were also calculated (Supplemental Table 1).
To estimate follow-up duration required to detect a treatment effect, we applied the Schoenfeld approximation assuming target hazard ratios of 0.5 and 0.1 (corresponding to 50% and 90% relative risk reductions, respectively), a two-sided α = 0.05, 80% power, and a total sample size of 200 participants with equal allocation between treatment and control groups. Monte Carlo simulations (5000 replicates) were performed by sampling from the pooled log-rate distribution using its estimated standard error and calculating follow-up time as the ratio of required events to the product of the simulated event rate and total sample size.19,20 Median and interquartile range (IQR) of required follow-up were reported for each endpoint to account for between-study heterogeneity.
Assumptions for power and detectable-effect calculations
Power calculations were conducted for a longitudinal slope-based endpoint representing the annualized rate of change in PR or QRS interval (ms/year). DM1 cardiac progression rates were informed by published longitudinal studies, with contemporary cohorts reporting PR and QRS rate progression ranging from 1.43–6.02 ms/year and 0.92–2.47 ms/yr, respectively.5,21,22 Because individual-level variance estimates were unavailable from the literature, variability was specified conservatively. The standard deviation (SD) of cumulative PR or QRS change over 5 years was assumed to be 10 ms, consistent with reported variability in longitudinal ECG studies. For shorter follow-up durations, the SD was scaled assuming approximately linear accumulation of variance over time (i.e., SD proportional to the square root of follow-up duration).
All calculations assumed α = 0.05, 80% power, equal allocation between treatment and control groups, and linear progression over time. The primary comparison was based on the difference in annualized progression rates between groups (treatment-by-time interaction). Treatment effects were defined as relative reductions in the progression rate compared with untreated disease, consistent with standard practice for disease-progression endpoints.
Report on the DMCRN/MDF cardiac endpoint workshop:
Topic I: the clinical impact of cardiac dysfunction in DM1
EP, cardiac rhythms in DM: arrhythmias and their impacts
William J. Groh, MD, MPH, Medical University of South Carolina, began the meeting with a broad overview of cardiac findings in the muscular dystrophies. Cardiac disease is a well-known complication of several muscle disorders, and for DM1, a range of cardiovascular defects have been reported. Individuals with DM1 are at risk for conduction abnormalities, arrhythmias, sudden death, and cardiac myopathy/heart failure, in order of decreasing prevalence. Cardiac causes have been shown to contribute to approximately 25–30% of deaths in individuals with DM1.
A DM1 arrhythmia registry, started in 1996, followed 406 genetically-confirmed individuals with DM1 over an 11-year follow up period. Importantly, they found that 65% of ECG's were abnormal in adult DM1 patients; 41.9% of individuals had first degree atrioventricular (AV) block, with prolongation of the PR interval (>200 ms), whereas 19.6% had an abnormal QRS duration. A Kaplan-Meier analysis revealed that most individuals with DM1 started experiencing cardiac events around ∼40 years of age. Additionally, the registry found that as age increases and CTG repeat length increases, the PR interval is longer. The registry data also showed that both clinical characteristics and ECG findings are independent predictors of cardiac disease. Atrial arrhythmia or ECG findings of severe conduction disease (e.g., PR > 240 milliseconds, 2nd/3rd-degree AV block, non-sinus rhythm) were predictive of sudden death.
Further review of the literature showed that other studies have found similar results – primary and meta-analyses show that 25–45% of individuals with DM1 have prolongation of the PR interval. Other ECG abnormalities such as bundle branch blocks and prolongation of the QRS interval (> 120 ms) have been shown to occur in 15–20% of cases.21,23,24 A systematic review that included 3677 individuals with DM1 across 13 studies found the mean prevalence of atrial fibrillation (AF) to be 10.9%. 25 Interestingly, a recent study by Austin et al. showed that AF and atrial flutter were present in 32.8% of their patients with DM1 who underwent annual 72-h ambulatory monitoring. 26 While this finding may be explained in part by ascertainment bias, as the patients were those referred to an electrophysiology clinic, it does suggest improved AF detection by extended cardiac rhythm monitoring over standard 12-lead ECG monitoring. More invasive electrophysiologic studies have also been performed in DM1, and generally, intracardiac measures correlate with surface measurements, with the exception that the His-Ventricular (HV) interval may be prolonged in a subset of individuals that have normal PR intervals on surface ECG. 27
Current treatment for the cardiac manifestations of DM1 is not disease specific. For patients that develop a reduced ejection fraction, guideline directed medical therapy includes ACE inhibitors or Angiotensin Receptor Blockers (ARBs), beta blockers, and aldosterone antagonists as well as angiotensin receptor-neprilysin inhibitors and SGLT2 inhibitors. If patients have atrial fibrillation or flutter, anticoagulation treatment is recommended as determined by validated stroke risk score. Symptomatic and prophylactic implantation of cardiac devices (e.g., cardioverter-defibrillator, pacemakers) have been studied as well, and have been found to decrease mortality for selected individuals with DM1. 28 Another medication commonly used for DM care, Mexiletine, a Class IB antiarrhythmic therapy, is clinically used to treat myotonia in DM1 by enhancing the fast inactivation of sodium channels. Importantly, randomized controlled trials between mexiletine and placebo have demonstrated no meaningful changes in PR, QRS, or QTc intervals over short-term follow-up, and in a larger 6-month trial, serial ECG and Holter monitoring likewise showed no effect on cardiac conduction measures compared with placebo.29,30
Overall, individuals with DM1 are at a high risk for conduction disease, arrhythmias, and sudden death. A smaller proportion of individuals have been found to have structural heart disease, further discussed in session iv. cardiac imaging.
Topic II: mechanism of cardiac dysfunction in DM
Pathophysiology of cardiac dysfunction in DM1
Thomas Cooper, MD, Baylor College of Medicine, presented a review of our current understanding of DM1 cardiac pathophysiology. It is well established that the core mechanism for DM1 is RNA toxicity, whereby DMPK mRNA transcripts with expanded CUG repeats form hairpins and sequester important splicing factors, such as muscleblind-like (MBNL) proteins, in nuclear foci. This functional depletion of MBNL causes global dysregulation of alternative splicing and other changes in RNA processing, resulting in hundreds of transcripts reverting to fetal isoforms. This ends in a heterogeneous array of clinical findings in which specific phenotypes, such as myotonia, heart block, or insulin resistance, are linked to splicing defects of certain genes, such as CLCN1 (chloride channel), SCN5A (sodium channel), or INSR (insulin receptor), respectively.
Several mis-spliced transcripts have been implicated in the cardiac phenotype of DM1. These include SCN5A, which encodes the cardiac voltage-gated sodium channel NaV1.5; RYR2 and ATP2A2 (SERCA2), which regulate calcium release and reuptake; and TNNT2, encoding cardiac troponin T. Additional splicing abnormalities have been observed in CAMK2D (a calcium/calmodulin-dependent kinase), KCNIP2, KCND3, and KCNAB1 (potassium channel subunits), as well as genes involved in cytoskeletal organization and signal transduction such as SORBS1, TMEM63B, DNM1L, GOLGA2, and FXR1. Presumably, the combined effect of these splicing abnormalities leads to impaired electrical conduction in the heart, delayed repolarization, disrupted excitation-contraction coupling, and impaired structural integrity of the myocardium.
Research regarding which mis-spliced events might be driving DM1 cardiac phenotypes is critical. From prior work in the field, we know that certain combinations of mis-spliced events is enough to recapitulate specific DM1 phenotypes. Cisco et al. showed that mice with forced skipping of exon 29 in the CaV1.1 calcium channel combined with loss of ClC-1 chloride channel function displayed markedly reduced lifespan, myotonia, weakness, impaired mobility, and reduced respiratory function. They also found that chronic administration of the calcium channel blocker, Verapamil, rescued survival and improved force generation, myotonia, and respiratory function. One of the priorities for the field is to recapitulate this study with other affected organ systems in DM1. Among these, SCN5A has emerged as a particularly interesting target. In experimental mouse models where exon 6A is forcibly included, optical mapping reveals a delay in atrial and ventricular conduction and repolarization. These mice exhibit left atrial and right atrial firing abnormalities and significant delays in ventricular depolarization compared to wild-type controls. However, CRISPR-based excision of exon 6A does not reverse the electrophysiological phenotype, suggesting that the problem extends beyond mis-splicing. Follow-up studies show that SCN5A transcript levels are significantly reduced in both DM1 human hearts and CUG960 transgenic mice. This points to a broader mechanism in which mis-splicing is potentially compounded by transcriptional downregulation or post-transcriptional instability of key cardiac transcripts.
DM1 cardiac disease involves other pathological features outside of cardiomyocyte splicing dysregulation. Histologically, DM1 hearts often exhibit selective degeneration of the conduction system, widespread interstitial and perivascular fibrosis, and focal fatty infiltration. A major objective is to understand how intrinsic defects, such as cardiomyocyte mis-splicing, interact with systemic features (i.e., autonomic nervous system dysfunction) and non-cardiomyocyte cells (i.e., fibroblasts, vascular smooth muscle, inflammatory cells) to contribute to DM1 cardiac disease. At a molecular level, key questions that need to be further addressed to definitively determine how the DM1 CTG repeat expansion affects cardiac function include:
What are the relative contributions of the cellular defects in the parenchyma versus the conduction system (i.e., delayed conduction, heart block)? Contributions of intrinsic defects in cardiomyocytes (i.e., dysfunctional ion channels, contractile proteins, signaling) versus tissue response (fibrosis and fatty replacement)? Contributions of the systemic features of DM1 to heart pathophysiology (i.e., autonomic nervous system dysfunction, vascular smooth muscle)? Contributions of non-cardiomyocytes to pathology (e.g., cardiac fibroblasts, inflammatory cells)?
Another proposed question in the field is, what drives the progression of cardiac disease in DM1? Although splicing abnormalities are present early, the clinical phenotype often worsens over time. The leading hypothesis is that somatic instability of the CTG repeat contributes to progressive worsening of disease. In tissues like the heart, where cellular turnover is low, the repeat expansion has a predisposition to lengthen over an individual's lifetime. To investigate this, Cooper and other colleagues have used the transgenic CUG960 mouse model that has an expanded repeat with interruptions, keeping the repeat length stable. These mice show a progressive cardiac phenotype, including increased heart mass, greater myocardial fibrosis, reduced ejection fraction, and longer QRS intervals with prolonged expression of the repeat, in the absence of somatic expansion. However, surprisingly, the degree of mis-splicing does not differ significantly between short- and long-term expression in this model. This suggests that disease progression may result from secondary changes—such as tissue remodeling, chronic stress signaling, or compensatory hypertrophy—rather than simply from increased burden of mis-spliced transcripts.
Topic III: measuring cardiac arrhythmias in clinical trials
Review of standard arrhythmia endpoints for consideration in clinical trials
Jordana Kron, MD, Virginia Commonwealth University, provided an overview of cardiac clinical trial endpoints and arrhythmia monitoring strategies, with a focus on their applicability to DM1. Establishing sensitive and reliable endpoints for progressive conduction disease and arrhythmias is essential for both clinical care and therapeutic trials in DM1.
ECGs remain a cornerstone of cardiac clinical assessment. They are simple, inexpensive, quick, and accessible tools that effectively measure cardiac rhythms. However, the utility of a standard ECG is limited by its inability to capture intermittent or paroxysmal arrhythmias, which are common in DM1. Holter monitors, which record cardiac rhythms continuously over 24–48 h, allow for more comprehensive and extended evaluation. They can detect atrial and ventricular arrhythmias, quantify burden of premature atrial contractions and premature ventricular contractions, assess variability in heart rate, and correlate patient-reported symptoms with arrhythmic episodes. Holter monitors can also distinguish between first-, second-, and third-degree heart blocks. Quantitative data derived from these monitors—such as arrhythmia burden and heart rate range—can be used to assess disease severity and progression, as well as overall autonomic tone.
For patients with infrequent or episodic symptoms, longer-term event monitors that record over 14 to 28 days offer a valuable alternative. These devices function similarly to shorter-term monitors but provide extended monitoring via patient-friendly adhesive patches. They often result in better patient adherence while still capturing arrhythmia data and facilitating symptom correlation. These monitors are useful in detecting intermittent AV blocks and atrial and ventricular arrhythmias that may not occur during a 48-h window.
Another long-term rhythm surveillance method is the use of implantable loop recorders (ILRs). These small devices, about the size of a flash drive, are inserted subcutaneously lateral to the sternum and can continuously record cardiac electrical activity for up to 4–5 years. ILRs can capture bradyarrhythmia, tachyarrhythmia, prolonged pauses, and patient-triggered events. Because they are implanted, they offer uninterrupted monitoring and high diagnostic yield, especially for arrhythmias that are rare, transient, or asymptomatic. Data from ILRs can be collected either synchronously, allowing for timely clinical intervention if needed, or asynchronously. These devices are also compatible with common imaging modalities, such as MRI and mammography.
Wearable technologies, such as smartwatches, have gained attention as potential tools for cardiac monitoring. Although they do not provide the resolution or accuracy of medical-grade diagnostics, they provide long-term, user-friendly access to rhythm data and may help detect gross abnormalities in heart rate or rhythm. While not currently suitable as standalone diagnostic tools in clinical trials, these technologies could serve as adjuncts for symptom monitoring, patient engagement, or exploratory endpoints.
Another option is pacemakers and implantable cardioverter-defibrillators (ICDs). These devices can continuously record atrial and ventricular activity, providing data in patients already receiving device-based therapies. ICDs offer the added advantage of therapeutic capability, delivering pacing or shocks in response to arrhythmias such as ventricular tachycardia (VT) or ventricular fibrillation (VF). Pacemakers do not have the same treatment capabilities. It was also noted during the workshop that not all individuals with DM1 qualify for or should receive device implantation, which limits the generalizability of this modality.
Results from clinical trials for other cardiac diseases were also discussed as potential models for the DM1 field. The CHASM study, led by Birnie et al., provided a template for monitoring subclinical conduction abnormalities, while the MAGiC-ART study highlighted the value of combining imaging, ECG, and extended rhythm monitoring in inflammatory cardiomyopathies.31,32 Importantly, these approaches are now beginning to be applied in DM1 clinical trials (NCT06844214). These efforts underscore the need to standardize cardiac endpoints and monitoring strategies in DM1, particularly as new disease-modifying therapies move into clinical testing.
Topic IV: the role of cardiac imaging and exercise in clinical trials
Using cardiac imaging in clinical trials
Greg Hundley, MD, Jennifer Jordan, PhD, MS, and Amy Ladd, PhD, Virginia Commonwealth University, gave an overview of CMR and the benefits it offers when assessing cardiomyopathy, particularly for muscular dystrophies such as DM1. CMR allows for quantitative analysis of both left and right ventricular function and provides tissue characterization, with or without gadolinium contrast. It can also evaluate other disease-relevant manifestations, including changes in vascular structures and body composition, making it pertinent for multisystemic diseases like DM1. Additionally, it is now a level 1 indication in cardiomyopathy screening.
Compared to echocardiography, CMR offers greater sensitivity and reproducibility in measuring structural changes. For example, detecting a 3% change in left ventricular ejection fraction requires far fewer subjects when using CMR (n = 15) compared to echocardiography (n = 102). Similarly, detecting a statistically significant 10-gram change in cardiac mass requires 9 participants with CMR versus 273 with echocardiography, highlighting its utility in clinical trial design for rare diseases, where statistical power may be limited.
Gadolinium-enhanced CMR provides both perfusion imaging and delayed enhancement imaging. First-pass perfusion highlights vascular defects, while late gadolinium enhancement (LGE) identifies areas of myocardial injury and replacement fibrosis. The pattern of enhancement often distinguishes ischemic from non-ischemic pathologies: endocardial-based fibrosis is generally ischemic, whereas mid-wall, epicardial, or diffuse enhancement is more typical of a non-ischemic processes such as sarcoidosis, myocarditis, or an infiltrative disease (e.g., cardiac amyloidosis). Despite this, LGE does not capture all forms of myocardial fibrosis—particularly interstitial fibrosis between myocytes. T1 and T2 mapping techniques improve sensitivity for diffuse changes, including edema, interstitial fibrosis, and early fibrotic changes not evident with LGE alone.
CMR also allows for assessment of vascular stiffness and body composition. For example, MR imaging can quantify fat distribution in the abdomen and in skeletal muscle.
From a research infrastructure standpoint, centralized CMR core labs can support large-scale, multi-center studies. Dr Jordan emphasized the importance of multisite harmonization, and having expertise regarding machine programming and image analysis. Prior efforts have processed datasets including thousands of echocardiograms and hundreds of CMR scans using standardized protocols across different scanner models, thus reducing the total number of participants required to detect a meaningful difference. Additionally, CMR offers additional benefits, such as no ionizing radiation, minimal contrast requirements, established outcome measures, and length of tests. A complete CMR scan without contrast takes approximately 8–12 minutes, while contrast-enhanced studies require around 30 minutes.
For greater context on the utility of CMR in DM1, a brief review was conducted of prior literature in the field using imaging technologies to gain insight into both functional and structural changes in DM1 hearts. Many of these studies have been smaller in scale than those utilizing surface ECG, and findings from echocardiography and doppler cohorts (n = 20–40 individuals) include wall motion abnormalities, decreased myocardial velocities and diastolic dysfunction in individuals with DM1 without any overt cardiac disease or changes in ejection fraction.33–36 CMR findings from select cohorts of patients with DM1 (n = 30–80 individuals) include decreased LV mass, increased T1 relaxation times, increased extracellular volume (ECV) fraction, decreased strain measurements, and increased late gadolinium enhancement (LGE), indicative of increased fibrosis and edema in the heart.37–43 As an example of the utility of a multimodal approach to identify cardiac involvement in DM1, a recent study in 195 individuals with DM1 found that the proportion of patients with cardiac involvement progressed from 42.1% at baseline to 65.6% after >10 years of follow-up. Standard ECG detected cardiac involvement in the majority of those affected, but ∼15% of patients had normal ECGs and abnormal Holter monitoring or echocardiograms. Further, presence of cardiac symptoms was relatively low (17%), underlying the importance of systematic work-up regardless of symptom status. 21
Moving forward: framework for defining clinical trial endpoints and directing future natural history study
To further develop a framework to assess cardiac outcomes for use as trial endpoints, we present an analysis based on two criteria: (1) correlation of outcomes with risk of cardiac mortality and morbidity in DM1 and (2) feasibility for incorporating into a clinical trial. The primary manifestation of cardiac involvement in DM1, and most studied, is conduction system disease and arrhythmia found in 30–75% of individuals with DM1 in cross-sectional studies. While these abnormalities range from mild PR prolongation (1st degree AV block) to severe findings (PR > 240 ms, QRS > 120 ms, non-sinus rhythm, ventricular tachyarrhythmia, or 2nd or 3rd degree AV block) on ECG, life-altering major cardiac events represent the target clinical outcomes for an experimental therapeutic.
To assess endpoint metrics of these manifestations, we first rate outcomes by how well they represent the risk of cardiac mortality and morbidity in DM1 (Figure 1, X-axis). The feasibility of outcome measures is then presented according to the reported annual incidence of events in at-risk DM1 populations (Figure 1, Y-axis; Supplemental Figure 1).
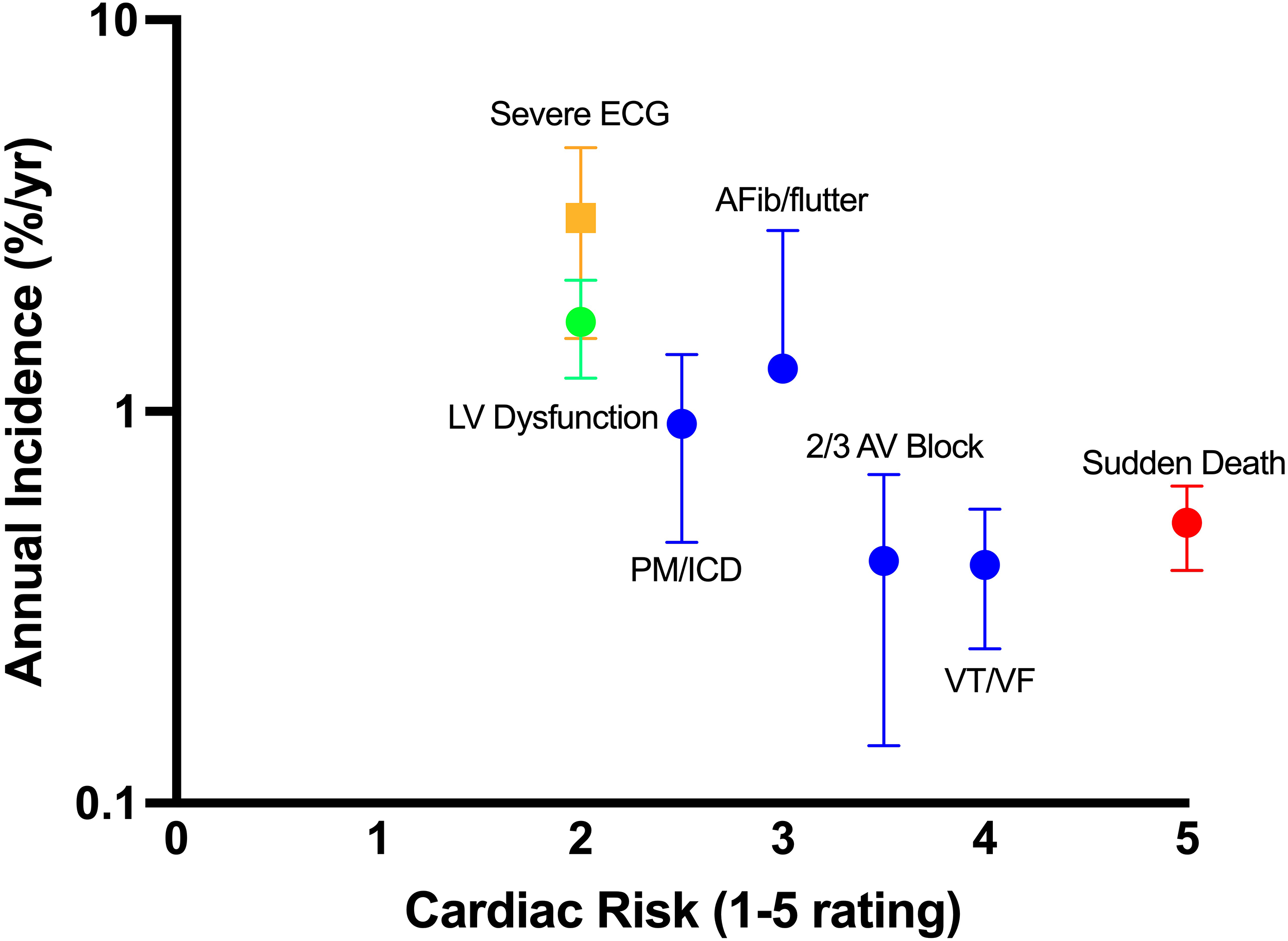
Assessment of cardiac outcomes by severity and incidence. Cardiac endpoints are rated 1 to 5 by expert judgement based on relative risk of mortality and morbidity against the reported annual incidence (log-scale). Values are reported as the mean ± SD.
Sudden death, life-threatening arrhythmia (VT/VF), ICD shocks, or decompensated heart failure represent the highest-risk cardiac manifestations in DM1 and are the most important core clinical outcomes for a therapeutic to prevent. However, low event frequency of these major cardiac events (<1% annual incidence) makes them less feasible for therapeutic trials. Sudden death or aborted sudden death (including appropriate ICD shocks or resuscitated cardiac arrest) occurs at an estimated pooled rate of 0.49% (range: 0.25–1.15%) per year in longitudinal DM1 cohort studies (Supplemental Table 1).5,11,21,23,44–46 Under these incidence assumptions, a time-to-event analysis indicates that, in a randomized trial of 200 participants, a therapy producing a 50% relative hazard reduction would require a median of 63.3 years (IQR 53.6-72.7) of follow-up to achieve adequate power to detect a treatment effect (Figure 2A). Even under a best-case scenario of a 90% relative hazard reduction, the required follow-up would be a median of 5.7 years (IQR 4.9-6.6) (Figure 2B).
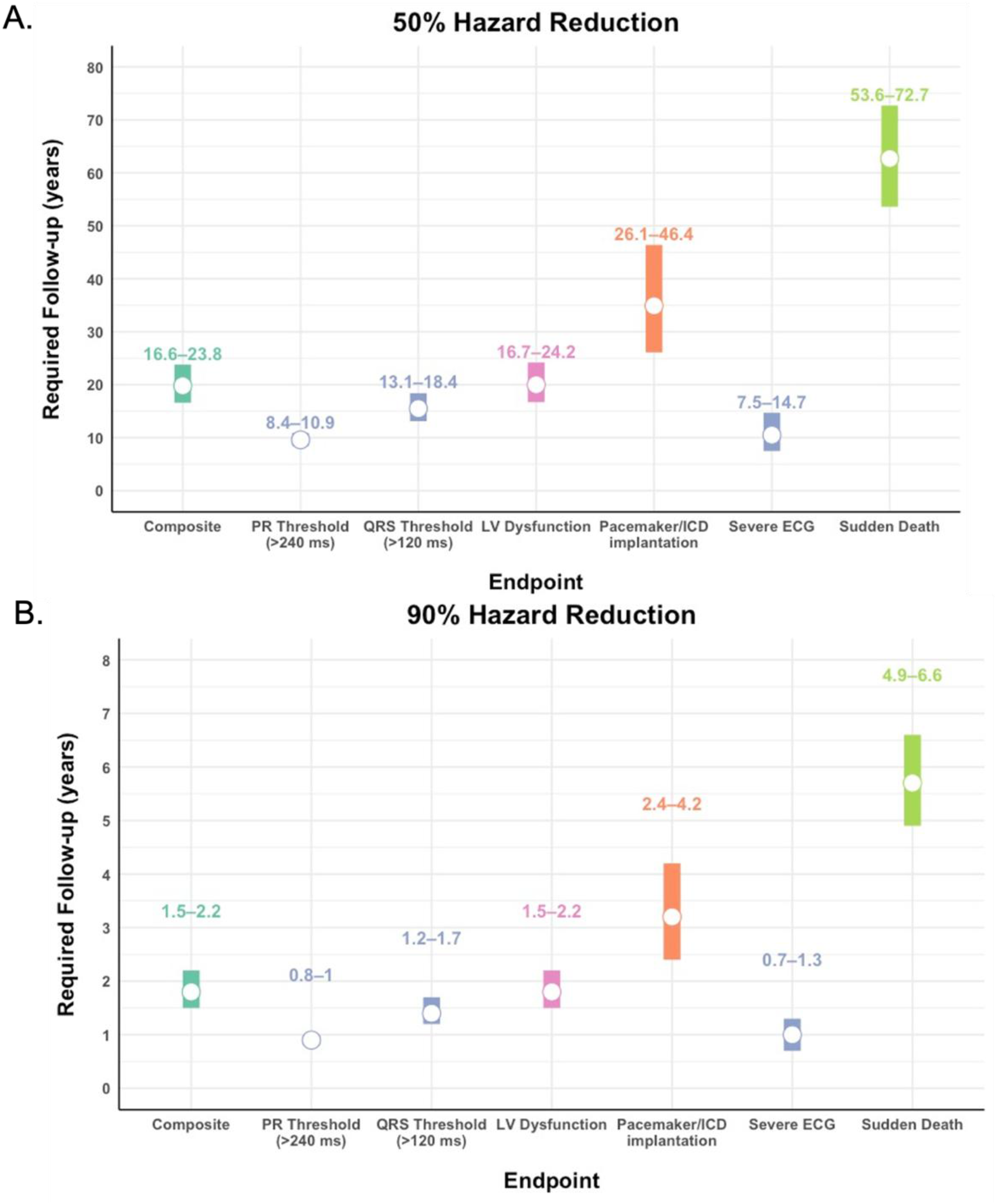
Required follow-up interval for DM1 cardiac endpoints. Interval plots showing the estimated trial duration (in years) required to detect a 50% (A) or 90% (B) hazard reduction for each outcome. Calculations assume n = 200 participants (100 per study arm; drug vs. placebo), α = 0.05, and 80% power, based on baseline incidence rates from longitudinal DM1 cardiac studies. Numbers above each bar represent the 25th–75th percentile range of required follow-up, and the white dot indicates the median. The composite endpoint includes sudden death, resuscitated cardiac arrest, ventricular tachyarrhythmias, atrial tachyarrhythmias, and 2nd/3rd degree atrioventricular block. Severe ECG changes include PR interval > 240 ms, QRS duration > 120 ms, and 2nd/3rd degree AV block.
Composite endpoints:
To attempt to shorten the expected trial time, we next consider composite endpoints that combine major cardiac events. For instance, in the study by Clementy et al., a composite endpoint of major bradyarrhythmic events was used to compare the utility of ECG versus in-lab electrophysiologic study for prognostication. 46 Similarly, for our analysis we considered an expanded arrhythmic event composite metric that includes sudden death, resuscitated cardiac arrest, ventricular tachyarrhythmias, atrial tachyarrhythmias, and 2nd/3rd degree atrioventricular block to look at feasibility of a composite clinical endpoint. The average incidence of the composite endpoint was 1.66% per year,5,22,46 although not all studies provided complete data for each metric so this likely represents an underestimate.
Nevertheless, time-to-event analysis suggests that a prolonged trial period with a median of 20.1 years (IQR 16.6-23.8) is still required, assuming the same trial size and a hazard reduction of 50%. For a more robust therapeutic effect of 90% hazard reduction, the median follow-up required is 1.8 years (IQR 1.5-2.2) (Figure 2).
Severe ECG abnormalities:
Given the limitation of major event frequency, another strategy is to use ECG conduction changes that are more common. Although variable in the literature, several ECG-based outcomes have been shown to portend increased risk of sudden death, including severe ECG changes (PR interval > 240 ms, QRS duration > 120 ms, 2nd/3rd degree AV block) and atrial arrhythmias. 5 Further, measurement of electrical cardiac activity either by surface ECG, event monitors, or implanted loop recorders is widely available and is commonly implemented/standard practice for clinical study. Using an incidence rate of 3.14% for severe ECG changes,5,21 a time-to-event analysis shows that it would take a median of 10.5 years (IQR 7.5-14.7) to detect a significant difference of a therapeutic that reduced the risk of ECG changes by 50% in a trial of 200 participants (Figure 2A), and a median of 1.0 year (IQR 0.7-1.3) follow-up is required for a therapeutic that reduced the risk of ECG changes by 90% (Figure 2B). One factor complicating the reported rates of cardiac conduction and rhythm disturbances is the sensitivity of surface ECG. Standard 12-lead surface ECG monitoring may fail to detect a substantial proportion of electrical abnormalities identified with extended ambulatory monitoring. In a longitudinal cohort, 29% of patients with normal 12-lead ECGs were found to have abnormalities on Holter monitoring over an 8-year follow-up period; however, the annual incidence of new Holter-detected abnormalities (3.5% per year) was comparable to that reported for ECG-based endpoints. 47 Similarly, EP studies have been used and studied in comparison to ECG at scale in France with the primary conclusion being that a proportion of patients with normal PR intervals on surface ECG are found to have delayed His-Ventricular (HV) conduction, and that using HV interval is a better prognostic marker of patients at high risk of having a major cardiac outcome.28,46 In both cases, although longer monitoring or EP studies identify a higher prevalence of abnormalities, the incidence of new cardiac events in at-risk individuals, i.e. meeting the surrogate outcome, is unchanged.
Continuous conduction measures:
As arrhythmogenic event frequency is low, an alternative to monitor progression of conduction disease would be to identify a continuous variable, such as PR interval and QRS duration, which has been studied extensively in multiple cohort studies. Using averaged rates of progression (PR: ∼2.98 ms/year; QRS: ∼1.63 ms/year),5,21,22 we estimated the minimum detectable difference in annualized progression for studies with varying sample sizes and follow-up durations.
For a study with 50 participants per group (n = 100 total), a 5-year follow-up would allow detection of slope differences of approximately 1.12 ms/year for PR and 1.12 ms/year for QRS, corresponding to ∼38% relative reduction for PR and ∼69% for QRS. Shortening follow-up to 3 years increases the detectable slope difference to 1.43 ms/year for PR and 1.43 ms/year for QRS, corresponding to ≥48% and ≥88% relative reduction, respectively. With only 2 years of follow-up, the study would be powered to detect only very large effects (≥59% reduction for PR, ≥107% reduction for QRS), limiting sensitivity of modest treatment effects (Table 1).
N = 100. Detectable annualized difference in PR/QRS progression by follow-up duration (50 participants per group, α = 0.05, 80% power).

Doubling the sample size to 100 participants per group (n = 200 total) improves sensitivity. For a 5-year follow-up, detectable slope differences are reduced to 0.80 ms/year for PR (∼27% relative reduction) and 0.80 ms/year for QRS (∼49% relative reduction). Detectable differences remain modest for 3 years of follow-up (1.03 ms/year; ∼35% for PR; 1.03 ms/year, ∼63% for QRS), and even 2-year studies can detect intermediate effects (1.25 ms/year, ∼42% for PR; 1.25 ms/year, ∼77% for QRS) (Table 2). Importantly, the longitudinal study by Groh et al., in line with the cardiac outcomes above, found the degree of PR interval and QRS duration prolongation was different between groups with mild or severe ECG changes, highlighting significant variation amongst the larger group, which would complicate their use as a clinical endpoint or, alternatively, suggests a target population for study.
N = 200. Detectable annualized difference in PR/QRS progression by follow-up duration (100 participants per group, α = 0.05, 80% power).

LV dysfunction and imaging endpoints:
In comparison to ECG changes, less has been published on longitudinal structural changes in DM1. Several larger longitudinal studies found the annual incidence of LV dysfunction to be 1.28–2.23% in the at-risk group.5,48 A time-to-event analysis indicates that, in a trial of 200 participants, detecting a therapeutic effect that reduces the risk of LV dysfunction by 50% would require a median of 20.0 years (IQR 16.7-24.2) of follow-up (Figure 2A). In contrast, a therapy that reduces the risk by 90% could be detected with a median of 1.8 years (IQR 1.5-2.2) of follow-up (Figure 2B). While this analysis suggests that a structural endpoint is similarly limited to those measuring arrhythmia endpoints, it is important to note that longitudinal studies thus far have only utilized echocardiography. Development of tissue fibrosis or more precise changes in pumping function using CMR has not been studied longitudinally.
Circulating cardiac biomarkers:
Last, few studies have examined blood biomarkers of cardiac injury and their relation to cardiac disease in DM1.49–54 In the study by Valaperta, et al., troponin-I and -T were measured along with NT pro-BNP in individuals with DM1. Only NT pro-BNP showed a modest correlation (r = 0.37) to cardiac conduction changes in this smaller cohort study (n = 60). Another group corroborated these findings, showing that while cardiac troponin I and CK-MB were elevated in their entire DM1 population compared to non-DM1 controls, NT pro-BNP and copeptin were specifically elevated in DM1 patients with atrial fibrillation. 54 Further longitudinal study would be required to better characterize the dynamics of these markers during cardiac disease progression and their utility as a biomarker of cardiac disease severity or progression.
In summary, we acknowledge that using publicly available datasets without patient-level data limits the precision of our estimates. Despite these limitations, our analysis does underscore one of the major takeaways from the workshop – the field would benefit from further longitudinal natural history studies of surrogate measures with higher sensitivity to capture both electrophysiologic and structural changes, such as monitoring conduction intervals with loop recorders and structural changes with CMR. These methods also allow for further evaluation of other less-studied outcomes and biomarkers that have been reported to be abnormal in DM1, such as heart rate variability,55,56 late potentials, 39 and changes in left ventricular mass.38,57 Also, while our understanding of the pathophysiology of cardiac manifestations in DM1 is incomplete, it likely represents a combination of progressive changes to tissue architecture (fibrosis/fatty replacement) and alterations in electrophysiologic function due to toxic RNA accumulation, and these studies may be designed to better understand the correlation of these factors. Last, based on our analysis, if the therapeutic benefit is not close to complete (i.e., 90% risk reduction), evaluation of major cardiac events is likely to require longer follow-up in a larger population, such as that seen with post-marketing surveillance studies.
Conclusion
In the era of systemic disease-modifying drugs for DM1, cardiac endpoints to assess disease severity and progression are crucial for designing clinical trials to evaluate novel therapeutics. The DMCRN/MDF Cardiac Workshop brought together basic scientists and clinical experts in the field to share data about current knowledge and brainstorm ideas for future directions. Here, we have supplemented that discussion with secondary analysis of the published literature on cardiac event rates and other biomarkers in DM1. Based on the workshop discussion and our analysis, we conclude that cardiac event frequencies are too low to be used as endpoints in standard clinical trials, and while monitoring progression of PR prolongation (slope of change) may be more feasible, this would still risk being underpowered to detect modest therapeutic effects. Due to these shortcomings, we propose further comprehensive natural history studies that would include longitudinal measurements of prolonged electrical monitoring, cardiac MRI, and serum markers, to yield the best combination of functional and structural information to identify specific cardiac endpoints and higher-risk study populations amenable to testing therapeutic efficacy.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602261433484 - Supplemental material for Developing endpoints for the cardiac burden in myotonic dystrophy type 1: A workshop report
Supplemental material, sj-docx-1-jnd-10.1177_22143602261433484 for Developing endpoints for the cardiac burden in myotonic dystrophy type 1: A workshop report by Julia M Hartman, Samuel Carrell, William J Groh, Thomas A Cooper, Jordana Kron, Greg Hundley, Jennifer Jordan, Amy Ladd, Man Hung, Nicholas E Johnson and on behalf of the Myotonic Dystrophy Clinical Research Network (DMCRN) and the Myotonic Dystrophy Foundation (MDF) in Journal of Neuromuscular Diseases
Footnotes
Acknowledgements
The authors would like to thank members of the DMCRN that contributed to the discussion, and Andy Rohrwasser, PhD from the myotonic dystrophy foundation for editorial comments.
Ethical considerations
Not applicable
Consent to participate
Not applicable
Consent for publication
Not applicable
Author contributions
Julia Hartman – Drafted the manuscript and performed the statistical analyses.
Samuel Carrell – Drafted and conceptualized the content of the manuscript.
William Groh - Presented at the workshop and edited the manuscript.
Thomas Cooper - Presented at the workshop and edited the manuscript.
Jordana Kron - Presented at the workshop and edited the manuscript.
Greg Hundley - Presented at the workshop and edited the manuscript.
Jennifer Jordan - Presented at the workshop and edited the manuscript.
Amy Ladd – Presented at the workshop and edited the manuscript.
Man Hung - Performed the statistical analyses.
Nicholas Johnson - Edited the manuscript and organized the workshop.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the FDA (7R01FD006071), Myotonic Dystrophy Foundation, Muscular Dystrophy Association, Takeda, Novartis, Pfizer, PepGen, Sanofi Genzyme, Vertex Pharmaceuticals, Arthex, Avidity, and Dyne Therapeutics.
Declaration of conflicting interests
Julia Hartman – none; Samuel Carrell – none; William Groh - Consultant agreements with Arrowhead Pharmaceuticals, Inc., Arthex Biotech, S.L; Thomas Cooper - none; Jordana Kron - none; Greg Hundley - none; Jennifer Jordan - none; Amy Ladd – none; Man Hung - none; Nicholas Johnson - has received grant funding from NINDS (R01NS104010, U01NS124974) and the FDA (7R01FD006071). He receives royalties from the CCMDHI and the CMTHI. He receives research funds from Novartis, Takeda, PepGen, Sanofi Genzyme, Dyne, Vertex Pharmaceuticals, Fulcrum Therapeutics, AskBio, ML Bio, and Sarepta. He has provided consultation for Arthex, Angle Therapeutics, Juvena, Rgenta, PepGen, AMO Pharma, Takeda, Design, Dyne, AskBio, Avidity, and Vertex Pharmaceuticals. He has equity in Repeat RNA Therapeutics, Angle Therapeutics, Evolyra Therapeutics, and Juvena Therapeutics.
Data availability
Not applicable
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.