
Abstract
Background:
Thymidine kinase 2 (TK2) deficiency is an ultra-rare, severe mitochondrial myopathy caused by pathogenic variants in TK2 and characterized by a wide range of ages at onset. The infantile form, presenting before 2 years of age, is the most rapidly progressive and is associated with a high risk of early mortality. We describe the clinical outcomes of early nucleoside therapy in a series of children with infantile-onset TK2 deficiency.
Methods:
We retrospectively reviewed four children with genetically confirmed infantile-onset TK2 deficiency treated with oral deoxycytidine/deoxythymidine (dC/dT) through an Early Access Program at two centers. Dosing was escalated to 800 mg/kg/day as tolerated. Patients were followed at baseline, Month 1, and regular intervals thereafter. Outcomes included neurological examinations, eight motor milestones, and respiratory and feeding support. Safety laboratory results, neuroimaging, and biopsy findings were reviewed.
Results:
Treatment began at 19–24 months (median duration 26 months; range: 4–81). All presented within the first year with hypotonia, motor regression, and respiratory and/or bulbar involvement. Two required invasive ventilation and three required tube feeding before therapy. After dC/dT initiation, all improved with no further milestone loss. Three achieved independent ambulation and stair climbing; the fourth, at 4 months of therapy, has begun unassisted walking. Both tracheostomized patients were weaned from ventilation, and enteral feeding was discontinued in all three within 1–6 months. Only mild dose-related diarrhea occurred in one patient.
Conclusion:
Early nucleoside therapy halts disease progression and restores motor function in infantile-onset TK2 deficiency, the most severe form of the disease.
Keywords
Introduction
Thymidine kinase 2 (TK2) deficiency (MIM# 609560) is a rare, life-threatening mitochondrial disorder caused by pathogenic variants in TK2, which encodes a mitochondrial deoxyribonucleoside kinase, essential for the mitochondrial nucleotide salvage pathway. 1 Loss of TK2 disrupts deoxypyrimidine phosphorylation, resulting in mitochondrial DNA (mtDNA) depletion or deletions and subsequent energy failure in metabolically active tissues such as skeletal and respiratory muscle.2,3 Since its first description in 2001, 4 approximately 150 patients have been reported worldwide.5–8
The infantile type of TK2 deficiency is characterized by symptom onset at or before 2 years of age and represents the most severe and rapidly progressive phenotype.7,9,10 Clinically, it may resemble spinal muscular atrophy (SMA) type 1, with profound hypotonia as a hallmark feature, commonly accompanied by decreased or absent reflexes.1,9 Early motor regression or impaired motor function is a characteristic feature of disease progression and may precede the development of overt bulbar, and respiratory weakness.1,10 Serum creatine kinase (CK) levels are frequently elevated but can be in the normal range in some patients. 9
Previous multicenter studies have demonstrated substantial clinical benefit of nucleoside supplementation in TK2 deficiency, including improved survival, stabilization of disease progression, and recovery of motor milestones.7,11 Here, we report a multicenter case series of four patients with infantile-onset TK2 deficiency treated with oral deoxycytidine/deoxythymidine (dC/dT) under a compassionate-use Early Access program, with a median treatment duration of 26 months (range: 4–81 months).
We observed rapid and significant functional recovery following early nucleoside treatment in all cases, which was sustained during long-term follow-up (≥18 months in 3 of 4 patients). The treatment was well tolerated, with only one patient developing moderate, self-limited diarrhea related to the medication. Our findings further reinforce the therapeutic impact of nucleoside supplementation in this rapidly progressive disorder. Our experience highlights the benefit of nucleoside therapy in the most severe form of TK2 deficiency and illustrates how timely intervention can alter the natural course of this devastating disease.
Materials and methods
Study design
We conducted a retrospective chart review of four children of Turkish origin with genetically confirmed TK2 deficiency who received oral deoxycytidine/deoxythymidine (dC/dT) therapy through an Early Access Program at two tertiary centers in Turkey. Genetic testing was performed at the participating centers. Segregation analysis of the identified TK2 variants was subsequently performed by Sanger sequencing in parental samples to confirm inheritance and variant phase. All eligible patients followed at the participating institutions during the study period were treated and included in this analysis; no untreated patients with comparable clinical characteristics were available for inclusion. The study was approved by the institutional review board, and written informed consent for participation and publication was obtained from the patients’ legal guardians.
Treatment protocol and follow-up
Oral deoxycytidine/deoxythymidine (dC/dT) was administered according to routine clinical practice. Treatment was initiated at a total dose of 260 mg/kg/day (130 mg/kg/day each of dC and dT, given three times daily) and, if tolerated, was escalated after 2 weeks to 520 mg/kg/day and after a further 2 weeks to a target maintenance dose of 800 mg/kg/day (400 mg/kg/day each).
Patients were evaluated at baseline, Month 1, every 3 months during the first year, and every 6 months thereafter. Assessments included physical examination (weight, height, vital signs, and neurological evaluation), documentation of motor milestone acquisition or loss, evaluation of respiratory and feeding status, and laboratory investigations. Eight predefined motor milestones were assessed: head control, independent sitting, standing with assistance, independent standing, assisted walking, independent walking, stair climbing with assistance, and independent stair climbing. Motor assessment was based on serial clinical neurological examinations together with documentation of motor milestone acquisition and loss. Evaluations included assessment of muscle tone, axial and proximal strength, deep tendon reflexes, and functional abilities at baseline and throughout follow-up. Respiratory status was evaluated based on the need for invasive or noninvasive ventilatory support and, when applicable, the daily duration of ventilation. Feeding status was classified as oral feeding or enteral support via nasogastric tube or gastrostomy, with documentation of feeding-related complications such as dysphagia.
Safety monitoring consisted of regular clinical assessments and routine laboratory testing, including creatine kinase, lactate, liver function tests (ALT, AST, total bilirubin, and GGT), renal function, and hematologic parameters, performed at scheduled visits according to standard clinical practice. CK monitoring varied between the two centers, and lactate levels were measured only at baseline and at the final visit. Brain MRI and muscle biopsy findings, when available as part of standard clinical evaluation, were reviewed and included in the analysis. Patients and caregivers were instructed on drug preparation and administration at treatment initiation and remained in regular contact with the treating team for monitoring and management of potential adverse events.
Data collection
Clinical data, including age at symptom onset, genetic results, motor milestones, feeding and respiratory status, and laboratory findings were reviewed. All available follow-up visits recorded during the study period were reviewed and included in the analysis. Because patients were seen every 3 months during the first year of treatment and every 6 months thereafter, parents were questioned about motor achievements that occurred since the previous visit, and these were recorded in the medical charts. These findings were captured through our retrospective chart review. Video recordings documenting motor function before and after treatment were available for all four patients and are provided as Supplementary Videos 1–4.
Results
Baseline clinical and laboratory findings
Table 1 shows clinical characteristics of the patients. All patients presented within the first year of life with hypotonia and delayed or regressed motor milestones, each losing at least one milestone before treatment (three patients lost ≥3). The maximal attained motor milestones and the ages at which they were gained and subsequently lost were: sitting unassisted (Patient 1, gained at 6 months, lost at 10 months), climbing stairs with support (Patient 2, gained at 15 months, lost at 19 months), walking with assistance (Patient 3, gained at 13 months, lost at 15 months), and independent walking (Patient 4, gained at 13 months, lost at 18 months). None regained lost milestones prior to treatment initiation.
Demographic, genetic, and clinical characteristics of four patients with TK2 deficiency treated with pyrimidine nucleoside supplementation.
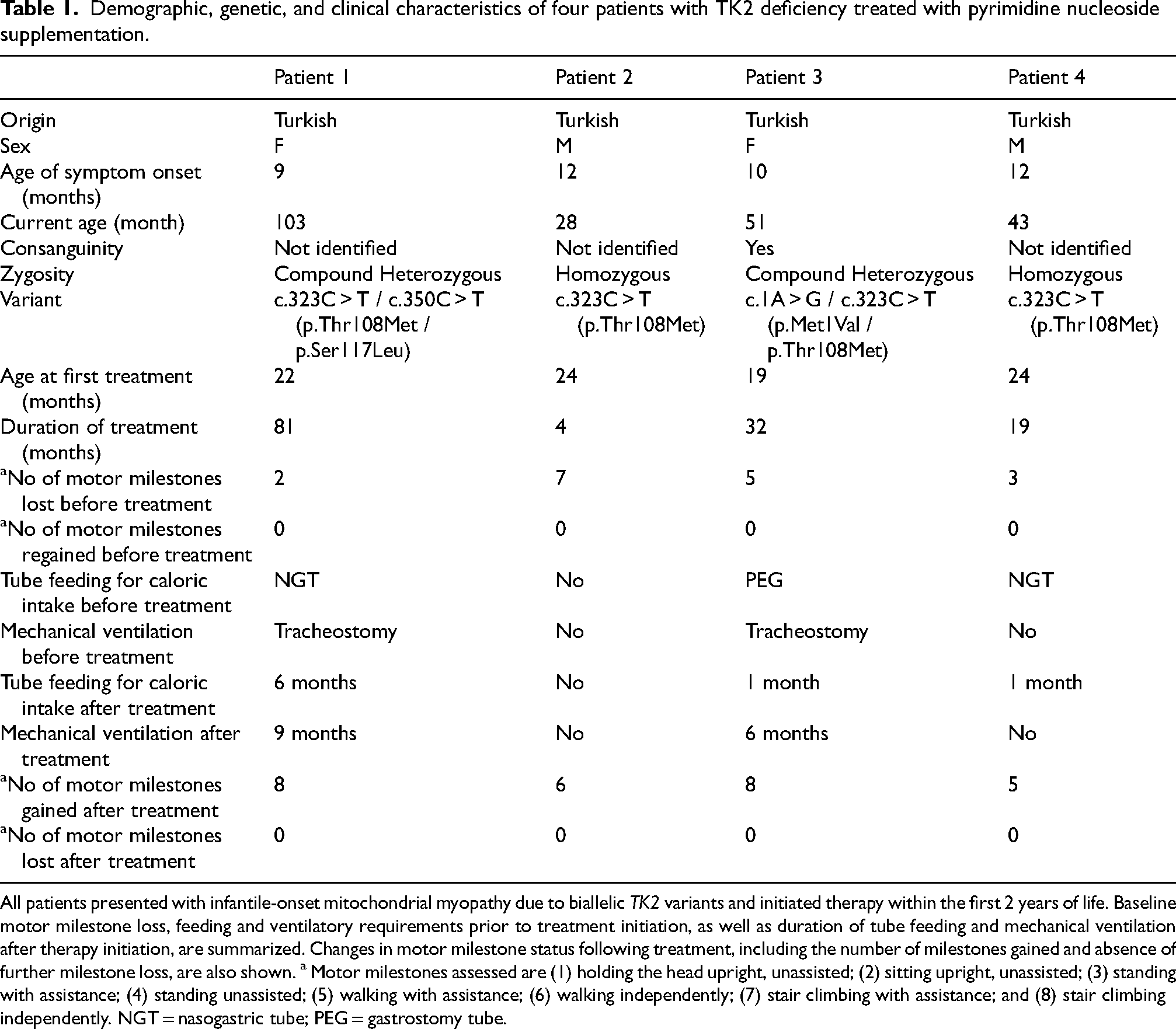
All patients presented with infantile-onset mitochondrial myopathy due to biallelic TK2 variants and initiated therapy within the first 2 years of life. Baseline motor milestone loss, feeding and ventilatory requirements prior to treatment initiation, as well as duration of tube feeding and mechanical ventilation after therapy initiation, are summarized. Changes in motor milestone status following treatment, including the number of milestones gained and absence of further milestone loss, are also shown. a Motor milestones assessed are (1) holding the head upright, unassisted; (2) sitting upright, unassisted; (3) standing with assistance; (4) standing unassisted; (5) walking with assistance; (6) walking independently; (7) stair climbing with assistance; and (8) stair climbing independently. NGT = nasogastric tube; PEG = gastrostomy tube.
Neurological examination consistently showed hypotonia, weakness of neck flexors and proximal limb muscles, and generalized hyporeflexia/areflexia. Feeding difficulties were observed in all cases; two patients required nasogastric tube feeding, and one required PEG. Three patients developed respiratory distress over time, and in two of these, tracheostomy and invasive ventilation were required.
Baseline clinical status at treatment initiation, including maximal motor function, neurological examination findings, feeding status and ventilatory support is summarized below for each patient.
Patient 1 was a 22-month-old girl at treatment initiation. The maximum motor milestone achieved prior to regression was independent sitting at 6 months (lost by 10 months); head control was acquired at 3 months and lost by 9 months. At treatment initiation, she had profound motor regression with complete loss of previously acquired milestones. Neurological examination showed severe axial hypotonia with marked axial and proximal weakness, reduced deep tendon reflexes, and only minimal antigravity limb movements; she was unable to lift her head in the supine position and could not pull to stand. Feeding difficulties developed at 13 months (4 months after symptom onset), requiring nasogastric tube support. Respiratory failure occurred at 18 months (9 months after symptom onset), leading to continuous invasive mechanical ventilation via tracheostomy (24 h/day) at treatment initiation.
Patient 2 was a 24-month-old boy at treatment initiation. Initial symptoms were observed at approximately 11–12 months, characterized by dysphagia to solids and recurrent vomiting. The highest motor milestone achieved was independent walking at 14 months, followed by stair climbing with assistance at 15 months; both abilities were lost by 19 months. At baseline, he had lost all previously acquired gross motor milestones and retained no functional motor skills. Neurological examination revealed severe axial hypotonia with prominent neck flexor and proximal limb weakness, along with reduced deep tendon reflexes. He was unable to lift his head in the supine position or to sit even with support. Distal strength was relatively preserved, with intact fine motor function. Dysphagia persisted at therapy initiation, restricting oral intake to liquids. He did not require tube feeding or ventilatory support at baseline.
Patient 3 was a 19-month-old girl at treatment initiation. The maximum motor milestone achieved was walking with assistance at 13 months, lost by 15 months. At treatment initiation, she had profound motor regression with loss of all previously acquired milestones. Neurological examination showed generalized hypotonia with marked neck flexor and proximal weakness and absent deep tendon reflexes; she was unable to lift her head against gravity. Gastrostomy feeding was initiated at 18 months (approximately 8 months after motor symptom onset). She required continuous invasive mechanical ventilation via tracheostomy (24 h/day) from 18 months of age.
Patient 4 was a 24-month-old boy at treatment initiation. The maximum motor milestone attained was independent walking at 13 months, lost by 18 months. At treatment initiation, he retained limited abilities including head control, independent sitting, and standing with assistance. Neurological examination demonstrated axial hypotonia with neck flexor and proximal weakness and absent deep tendon reflexes. He developed tachypnea at 21 months and received intermittent supplemental oxygen, without need for invasive or noninvasive ventilatory support. Feeding difficulties began at 23 months (approximately 11 months after motor symptom onset), necessitating nasogastric tube feeding.
Baseline CK levels were elevated in all patients, ranging from 415 to 4653 U/L. Lactate levels were elevated in all (Patient 1: 63 mg/dL, Patient 2: 47.2 mg/dL, Patient 3: 33.7 mg/dL, Patient 4: 29.7 mg/dL, reference range: 4.5–19.8 mg/dL).
Brain MRI was unremarkable in all cases. A muscle biopsy from Patient 1 showed features consistent with mitochondrial myopathy.
Genetic testing identified the recurrent TK2 variant c.323C > T (p.Thr108Met) in all four patients, present in homozygous form in Patients 2 and 4, and in compound heterozygous form in Patients 1 and 3 in combination with c.350C > T (p.Ser117Leu) or c.1A > G (p.Met1Val). The c.323C > T (p.Thr108Met) and c.350C > T (p.Ser117Leu) variants were classified as pathogenic, while c.1A > G (p.Met1Val) was classified as likely pathogenic according to ACMG guidelines. 12 Parental consanguinity was present in one family.
Segregation analysis confirmed parental origin of the identified TK2 variants in all patients. In Patients 1 and 3, the two variants were inherited from different parents, consistent with compound heterozygosity in trans (Patient 1: father c.323C > T [p.Thr108Met], mother c.350C > T [p.Ser117Leu]; Patient 3: mother c.1A > G [p.Met1Val], father c.323C > T [p.Thr108Met]). In Patients 2 and 4, who carried homozygous variants, both parents were heterozygous carriers.
Response to treatment
All patients began therapy at ≤24 months of age (22, 24, 19, and 24 months; Tables 1 and 2), corresponding to symptom durations from disease onset to treatment initiation of 13, 12, 9, and 12 months, respectively.
Longitudinal biochemical, motor, respiratory, and feeding outcomes following pyrimidine nucleoside therapy in four patients with TK2 deficiency.
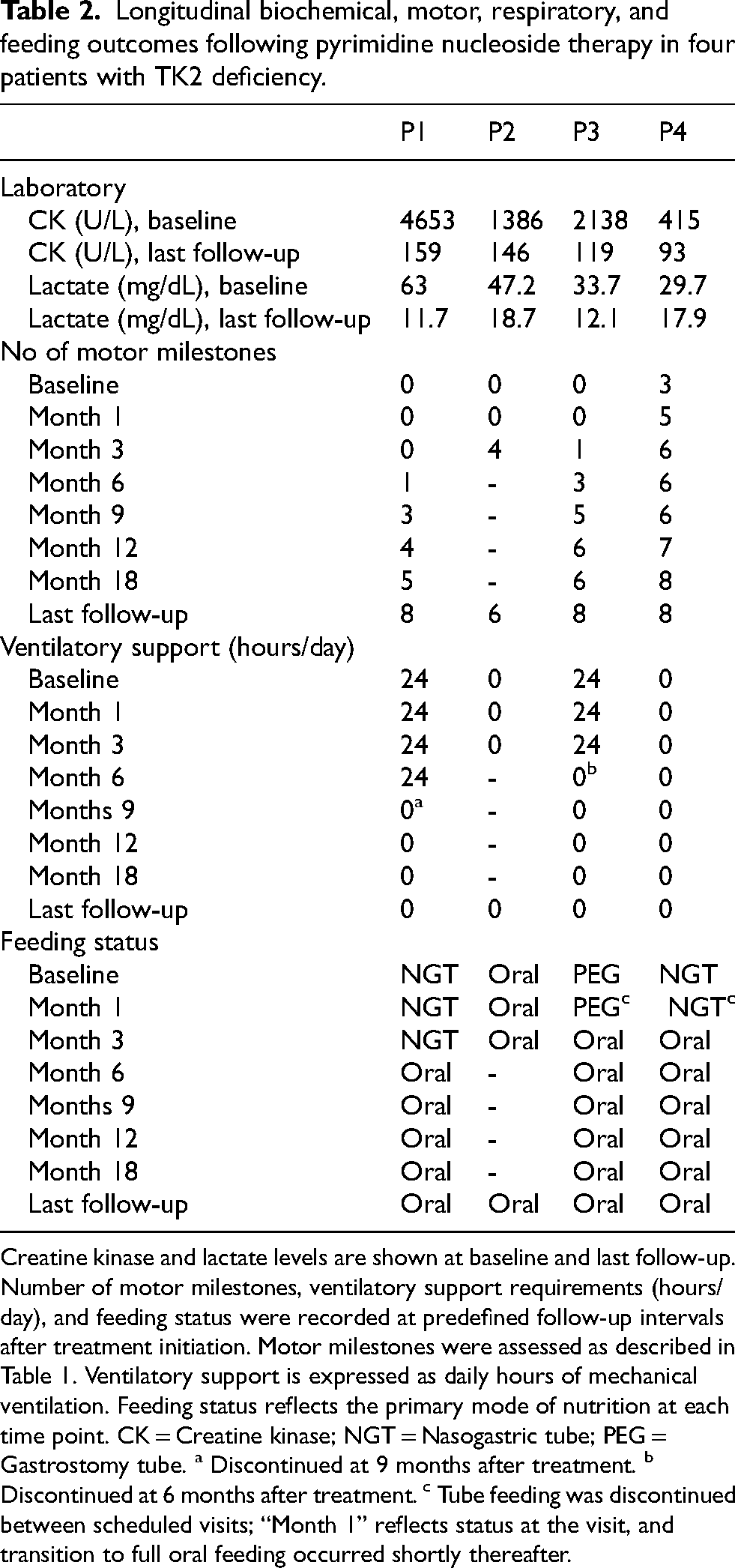
Creatine kinase and lactate levels are shown at baseline and last follow-up. Number of motor milestones, ventilatory support requirements (hours/day), and feeding status were recorded at predefined follow-up intervals after treatment initiation. Motor milestones were assessed as described in Table 1. Ventilatory support is expressed as daily hours of mechanical ventilation. Feeding status reflects the primary mode of nutrition at each time point. CK = Creatine kinase; NGT = Nasogastric tube; PEG = Gastrostomy tube. a Discontinued at 9 months after treatment. b Discontinued at 6 months after treatment. c Tube feeding was discontinued between scheduled visits; “Month 1” reflects status at the visit, and transition to full oral feeding occurred shortly thereafter.
After therapy initiation, no patient lost previously acquired milestones, and all regained at least two milestones that had been lost before treatment. Three patients achieved independent ambulation, and the fourth, currently 4 months into therapy, has recently begun unassisted walking. Both patients who required invasive ventilation (Patients 1 and 3) were successfully weaned from tracheostomy after 9 months and 6 months of therapy, respectively, with no further need for respiratory support. Enteral feeding via nasogastric or gastrostomy tube was discontinued in all three supported patients within 1–6 months of treatment initiation. Table 2 summarizes longitudinal biochemical (CK and lactate) and clinical outcomes (motor milestones, respiratory support, and feeding status) across predefined follow-up visits. Motor milestone trajectories before and after treatment initiation are shown in Figure 1.
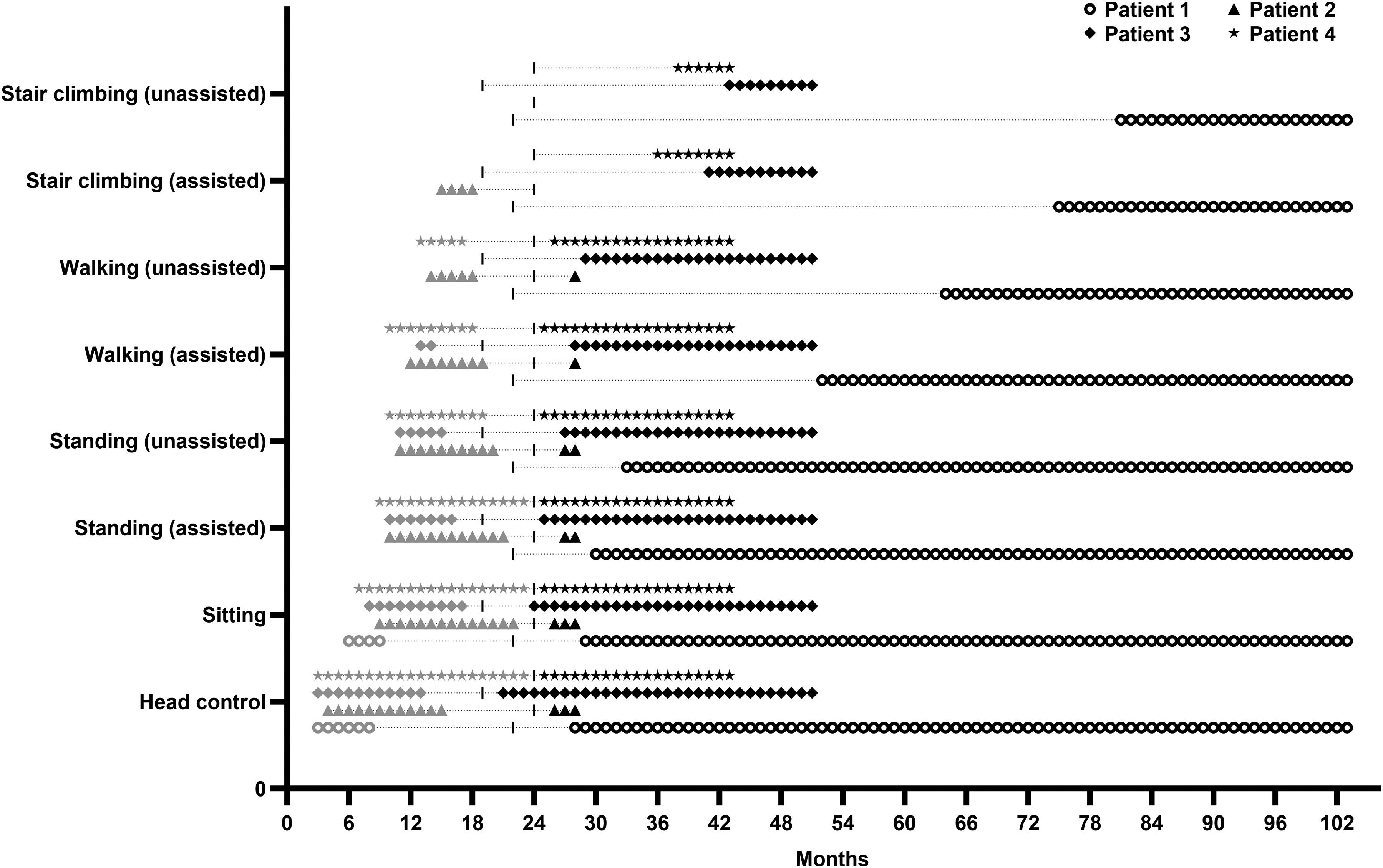
Longitudinal motor milestone trajectories following pyrimidine nucleoside therapy in infantile-onset TK2 deficiency. Timing of acquisition, loss prior to treatment, and subsequent recovery of eight predefined motor milestones is shown for four patients. Milestones include head control, independent sitting, standing with assistance, standing independently, walking with assistance, independent walking, stair climbing with assistance, and independent stair climbing. Symbols indicate individual patients (Patient 1–4), and the vertical dashed line (I) denotes the age at treatment initiation for each patient. Pretreatment visits are shown with gray and posttreatment visits are shown with black symbols.
Patient 1 improved within weeks, regaining head control and independent sitting at 6 and 7 months of therapy. She attained assisted walking at month 30 and independent ambulation by month 42, later achieving independent stair climbing. At last follow-up (81 months after treatment initiation), she remained ambulatory with a wide-based gait, moderate axial/proximal weakness, and reduced reflexes. She remained free of respiratory support after month 9, and full oral feeding was re-established by 6 months; the nasogastric tube was retained solely for medication administration until month 57 of therapy.
Patient 2, currently in the early phase of therapy (4 months), has demonstrated motor improvement, including regained head control, independent sitting, and recent onset of unassisted walking, with partial improvement in axial and proximal weakness, with hyporeflexia. Dysphagia has also improved.
Patient 3 showed clinical improvement within the first month, transitioning to full oral feeding with discontinuation of PEG support. She regained head control and independent sitting at months 2 and 5, progressed to assisted walking by month 9, and achieved independent ambulation by month 10. Tracheostomy was closed after 6 months of therapy. At last follow-up (32 months after treatment initiation), she was able to climb stairs independently (achieved at month 24), with only very mild residual neck and proximal lower limb weakness and reduced reflexes.
Patient 4 transitioned to full oral feeding within the first month, regained standing and assisted ambulation early in therapy, and achieved independent walking by month 2. Independent stair climbing was attained by month 14. At last follow-up (19 months after treatment initiation), he had only mild residual proximal weakness and reduced reflexes.
These functional gains are further demonstrated in Supplementary Videos 1–4.
Treatment was overall well tolerated. At the last follow-up (4–81 months after treatment initiation), all patients were clinically stable, orally fed, and free from ventilatory support. Reductions were observed in serum CK levels (93–159 U/L) and in lactate levels (11.7–18.7 mg/dL) at the last visit. One patient (Patient 2) developed dose related, mild/moderate diarrhea but did not require treatment interruption or dose adjustment.
Discussion
Our observations in these four children with infantile-onset disease demonstrate that early initiation of nucleoside therapy effectively halts disease progression and leads to rapid, sustained improvements in motor, respiratory, and feeding functions. All patients who began treatment within two years of age exhibited remarkable recovery of motor milestones, allowing discontinuation of mechanical ventilation and enteral feeding support.
Age at onset is an important determinant of disease severity in TK2 deficiency. 7 The disease presenting ≤2 years of age is the most severe form and is characterized with developmental regression due to rapidly progressive myopathy, and early respiratory failure. Infantile onset disease often fatal within the first two years.9,10 Until recently, management of TK2 deficiency was limited to supportive care.
Pyrimidine nucleoside replacement therapy with exogenous deoxycytidine and deoxythymidine has emerged as a targeted therapeutic approach for TK2 deficiency. These substrates enable mtDNA replication through the activity of cytosolic enzymes, thymidine kinase 1 (TK1) and deoxycytidine kinase (dCK), alongside any residual mitochondrial TK2 function. 13 Accumulating evidence indicates that pyrimidine nucleoside therapy can restore mitochondrial nucleotide balance, promote recovery of lost motor milestones, stabilize disease progression, and prolong survival.7,11,13–15 After reviewing data from two retrospective chart review studies (NCT03701568 and NCT05017818), an open label phase 2 study (NCT03845712) of continuation treatment in subjects participated in the retrospective study MT 1621-101 (NCT03701568) and others treated nucleos(t)ide treatment, and expanded access programs (NCT06590493), in November 2025, the U.S. Food and Drug Administration (FDA) approved Kygevvi™ (doxecitidine and doxribtimine) as the first therapy for TK2 deficiency.
These recently published multicenter retrospective studies leading to the approval of nucleoside treatment for TK2 deficiency provide important context for our early-treated cohort. In the largest published series of pyrimidine nucleos(t)ide therapy in TK2 deficiency (n = 38, eight centers), approximately 40% of patients had symptom onset within the first two years of life, and treatment was associated with improved survival compared with untreated historical cases, although therapy was frequently initiated late (often >10 years after onset; median exposure 1.6 years). 7 Our report complements these findings by focusing on uniformly early therapy, with all patients starting within the first two years of age after only 9–13 months of disease duration and follow-up of 4–81 months. In that multicenter cohort, nucleos(t)ide supplementation was linked to clinical stabilization, with no further motor milestone loss after treatment initiation and recovery of previously lost abilities in a substantial proportion of pediatric-onset patients. Respiratory and bulbar outcomes likewise showed stabilization or improvement, including discontinuation of ventilatory or feeding support in some individuals, and no new requirement for ventilation among previously unsupported patients during therapy. Earlier compassionate-use data also support benefit in severe infantile-onset TK2 deficiency. In a treated series of 16 patients, five with rapidly progressive early-onset severe myopathy showed clinically meaningful motor improvement and stabilization of ventilatory and feeding needs. 11 Biomarker studies also suggest an age-dependent response, with the greatest clinical benefit observed in pediatric patients, supporting the concept of an early therapeutic window. 16 Consistent with these reports, none of our patients lost additional milestones after treatment initiation, all regained previously lost motor functions, and all four achieved independent ambulation. Notably, both invasively ventilated infants in our cohort were successfully weaned from tracheostomy, and all patients achieved full oral feeding, remaining ventilator-free and orally fed at last follow-up.
A consistent genotype–phenotype correlation has not been established in TK2 deficiency, apart from selected recurrent variants. 1 All four patients in our cohort carried the TK2 variant c.323C > T (p.Thr108Met) in either homozygous or compound heterozygous form. Previously published case reports and series suggest that p.Thr108Met is among the most recurrent TK2 alleles and has been reported across infantile-, childhood-, and late-onset phenotypes, with outcomes ranging from rapidly progressive early disease to milder adult presentations, underscoring substantial clinical heterogeneity.10,17–21 In treatment cohorts, p.Thr108Met has been observed in individuals with severe early-onset disease requiring invasive ventilation and tube feeding at baseline, with variable degrees of improvement in motor milestones, respiratory status, and bulbar/feeding functions following pyrimidine nucleoside therapy. 11 In addition to p.Thr108Met, two of our patients carried p.Ser117Leu or p.Met1Val in compound heterozygosity. These variants have not been frequently reported among the most recurrent pathogenic TK2 alleles, supporting their relative rarity and the importance of continued variant-stratified outcome reporting. Our uniformly early treated cohort therefore provides additional long-term evidence of sustained milestone stabilization and recovery within this recurrent genotypic context.
This study has several limitations. The retrospective design, small sample size, and absence of an untreated control group are important limitations of this study. However, these constraints likely reflect current clinical practice in most centers, as patients with genetically confirmed TK2 deficiency typically receive pyrimidine nucleoside therapy following diagnosis in this rare and potentially life-threatening disorder. Standardized functional motor scales were not applied systematically during follow-up, as patients were treated under an Early Access Program with monitoring focused on motor milestones, feeding status, and respiratory support; thus, the retrospective nature of data collection precluded uniform functional assessment across visits. Consequently, longitudinal outcome evaluation relied primarily on structured neurological examination and clinically meaningful functional milestones. In addition, growth differentiation factor-15 (GDF-15), a proposed biomarker of disease severity and treatment response in TK2 deficiency, 16 was not routinely measured in this cohort, limiting biomarker-based correlation with clinical outcomes during follow-up.
In conclusion, clinicians should keep in mind TK2 deficiency within the differential diagnosis of early onset SMA. Our findings provide further evidence that early initiation of pyrimidine nucleoside therapy can dramatically alter the natural course of infantile-onset TK2 deficiency, the most severe form of the disease. These results highlight the transformative potential of timely therapeutic intervention in patients with TK2 deficiency.
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Footnotes
Acknowledgment
We sincerely thank the patients and their families for their participation. Deoxycytidine and deoxythymidine were supplied through a UCB Early Access Program. We acknowledge UCB for providing the study medication.
Ethical statement
This study follows the principles of the Declaration of Helsinki.
Author contributions
GA collected clinical data and drafted the manuscript. JS contributed to patient follow-up and data collection. HT is the principal investigator for the study and reviewed and supervised the study and critically revised the manuscript. All authors read and approved the final manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.