
Abstract
Background:
The number of mutations in nuclear encoded genes causing mitochondrial disease is ever increasing. Identification of these mutations is particularly important in the diagnosis of neuromuscular disorders as their presentation may mimic other acquired disorders.We present a novel heterozygous variant in mitochondrial fission factor (MFF) which mimics myasthenia gravis.
Objective:
To determine if the MFF c.937G>A, p.E313K variant causes a mild mitochondrial phenotype.
Methods:
We used whole exome sequencing (WES) to identify a novel heterozygous variant in MFF in a patient with ptosis, fatigue and muscle weakness. Using patient derived fibroblasts, we performed assays to evaluate mitochondrial and peroxisome dynamics.
Results:
We show that fibroblasts derived from this patient are defective in mitochondrial fission, despite normal recruitment of Drp1 to the mitochondria.
Conclusions:
The MFF c.937G>A, p.E313K variant leads to a mild mitochondrial phenotype and is associated with defective mitochondrial fission in patient-derived fibroblasts.
INTRODUCTION
Mitochondrial fission factor (MFF, OMIM# 614785) is a novel gene associated with mitochondrial disease. It is an adaptor protein for Drp1 and is involved in fission events in both mitochondria as well as peroxisomes, and it is a regulator of peroxisome maturation [1, 2]. In mitochondria it functions by recruiting Drp1 to active fission sites on the mitochondrial membrane during homeostatic fission as well as during mitophagy [1, 4]. MFF was initially identified through an siRNA screen of mitochondria morphology factors in Drosophila Ds2R+ cells [1]. It was found to localize to the mitochondria, anchored to the outer mitochondrial membrane. It also co-localizes to peroxisomes, although to a lesser degree. MFF preferentially recruits the oligomerized form of Drp1 [5]. MFF has also been shown to play a role in re-perfusion injury in cardiac tissue [6, 7]. In cancer it is needed for mitochondrial fragmentation and apoptosis [8, 9]. Over-expression of MFF in fibroblasts leads to mitochondrial fragmentation and metabolic reprogramming [10].
Recessive mutations in MFF have been reported in infants with a presentation similar to Leigh-syndrome [11–13]. The symptoms are severe and can include encephalopathy as well as myopathy. Surprisingly a defect in mitochondrial respiratory chain is not necessarily part of the pathogenic mechanism, as activities of mitochondrial respiratory chain complexes from muscle tissue were normal in two patients [12]. Nonetheless, the morphology of mitochondria and peroxisomes was affected. Mitochondria were elongated, with increased branching and abnormal distribution of Drp1.
In the current study, we present a case of late onset mitochondrial phenotype associated with a heterozygous c.937G>A, p.E313K variant in MFF. A muscle biopsy revealed an increased number of COX negative fibers suggestive of mitochondrial disease. We suggest that patients heterozygous in mutations in MFF can present with a mild mitochondrial phenotype which may mimic myasthenia gravis.
MATERIALS AND METHODS
Patient recruitment
The index patient was seen at the Johns Hopkins Hospital School of medicine. All subjects received clinical evaluations, genetic testing and skin biopsies at the National Institutes of Health (NIH) in Bethesda, MD under IRB-approved protocol 00-N-0043 “Clinical and Molecular Manifestations of Inherited Neurological Disorders.” Written informed consent was received before inclusion in the study.
Mitochondrial respiration
Mitochondrial OCRs were measured by using an XF96 Extracellular Flux Analyzer (Seahorse Bioscience) [14, 15]. Cells were seeded at 5,000 cells/well in an XF 96-well culture microplate and cultured for 24 h. Cells were washed twice in XF base medium supplemented with 25 mM glucose and 4 mM L-glutamine. The culture medium was replaced with the XF base medium and then cells were incubated at 37°C in a CO2-free incubator for 1 h. OCR measurement was performed according to the manufacturer’s instructions. Baseline OCR was recorded three times, and then, 2μM oligomycin, 1μM FCCP, and 0.5μM rotenone/antimycin A were sequentially injected into each well. OCRs were normalized relative to the amount of protein in each well. The first measurement value of OCR in control C was set to 1.
Whole exome sequencing
Genomic DNA was isolated from 10 mL of whole blood by isopropanol precipitation and washed with 70% ethanol. DNA was subsequently cleaned up with phenol/chloroform extraction and sodium acetate precipitation. After again washing with 70% ethanol, the DNA pellet was allowed to air dry for 5 minutes, and then resuspended in 10 mM Tris, 0.1 mM EDTA, pH7.8 to a concentration of 100 ug/ml. The NimbleGen SeqCap EZ Version 3.0 + UTR capture kit was used to cover 96Mb. Sequencing was performed at the NIH Intramural Sequencing Center (NISC). Illumina lanes of the captured DNA were sequenced to provide coverage to call MPG genotypes with a score of 10 in at least 85% of targeted bases. Reads were mapped to NCBI build 37 (hg19) using the Illumina aligner “ELAND” (Efficient Large-scale Alignment of Nucleotide Databases (Illumina, San Diego, CA). When at least one read in a pair mapped to a unique location in the genome, that read and its pair were then subjected to a more accurate alignment with Novoalign V3.02.07. The aligned lane bam files were merged, sorted, and indexed. Duplicate sequence reads derived from the same original DNA molecule, a PCR artifact characterized by molecules having the exact same alignment coordinates for both Read 1 and Read 2, were removed with Samtools. These alignments were stored in BAM format, and then fed as input to bam2mpg (http://research.nhgri.nih.gov/software/bam2mpg/index.shtml), to call genotypes at all covered positions using a probabilistic Bayesian algorithm (Most Probable Genotype, or MPG). Genotype calls were compared against Illumina Human 1M-Quad genotype chips, and genotypes with MPG score of 10 or greater show >99.89% concordance with SNP Chip data. Sequence bases with Phred quality score <20 (Q20) were ignored. Only reads with mapping quality >30 were included in the analysis. Sequence variant calls were evaluated using VarSifter software (NHGRI) and filtered using filter sets for myopathy and nuclear encoded mitochondrial genes curated through literature review. The ExAC Browser (Broad Institute) was used to evaluate allele frequency and confirmatory sequencing was performed by GeneDx (Gaithersburg, MD).
Cell culture
Human fibroblasts were cultured in Dulbecco’s Modified Eagle Medium containing 10% fetal bovine serum. WT and MFF–KO MEFs have been previously described [4]. MEFs were cultured in Iscove’s modified Dulbecco’s medium containing 10% fetal bovine serum [16].
Plasmids
Human MFF isoform1 (www.uniprot.org) was PCR-amplified from total cDNA derived from HEK293T cells using the following primers, 5’-TAGGGATCCGCCACCATGAGTAAAGGAACAAGCAGTGACACATCAC-3’ and 5’-TAGCTCGAGCTAGCGGCGAAACCAGAGCCAG-3’, and then cloned into BamHI and XhoI sites of the pcDNA3.1 plasmid. The MFF (E313K) plasmid was generated by replacing the guanine with adenine (937G>A) in the MFF (WT) plasmid. First, two partial fragments of MFF were PCR-amplified from the MFF (WT) plasmid using the following primers, 5’-TAGGGATCCGCCACCATGAGTAAAGG-3’ and 5’-CTCTTTTAGCACGTTCTTTGTTCTTCTC-3’, and 5’-GAACAAAGAACGTGCTAAAAGAGAAATGGTC-3’ and TAGCTCGAGCTAGCGGCGAAACC-3’. An underlined case is the added mutation. Second, full length of MFF with E313K variant was PCR-amplified from these two products of the first PCR using the following primers, 5’-TAGGGATCCGCCACCATGAGTAAAGG-3’ and 5’-TAGCTCGAGCTAGCGGCGAAACC-3’, and then cloned into BamHI and XhoI sites of the pcDNA3.1 plasmid.
Immunofluorescence microscopy
Human fibroblasts and MEFs were seeded in 8-well chambered coverglasses and cultured at 37°C for 24 h. Human fibroblasts were incubated with 10μM FCCP or DMSO at 37°C for 30 min. MEFs were transfected with plasmids using Lipofectamine 2000 (11668019, Invitrogen) overnight. Cells were fixed in pre-warmed (37°C) PBS containing 4% paraformaldehyde for 20 min, washed three times in PBS, permeabilized with PBS containing 0.1% Triton X-100 for 8 min, washed again three times in PBS, and blocked in PBS containing 0.5% BSA at room temperature for 30 min (Adachi et al., 2016). Cells were then incubated with primary antibodies in PBS containing 0.5% BSA at 4°C overnight. The antibodies used were PDH (1:400 in PBS containing 0.5% BSA, ab110333, Abcam), Pex14 (1:1,000 dilution, 10594-1-AP; Proteintech), Tom20 (1:300 dilution, sc-11415; Santa Cruz Biotechnology), Drp1 (1:300 dilution, 611113; BD Biosciences) and MFF (1:200 dilution, HPA010968; Sigma-Aldrich). Cells were washed three times in PBS and incubated with appropriate secondary antibodies at room temperature for 1 h. Cells were washed again, three times in PBS. The samples were observed using an LSM800 GaAsP laser scanning confocal microscope (Kageyama et al., 2014; Yamada et al., 2016). Image analysis was performed using ImageJ software.
Western blotting
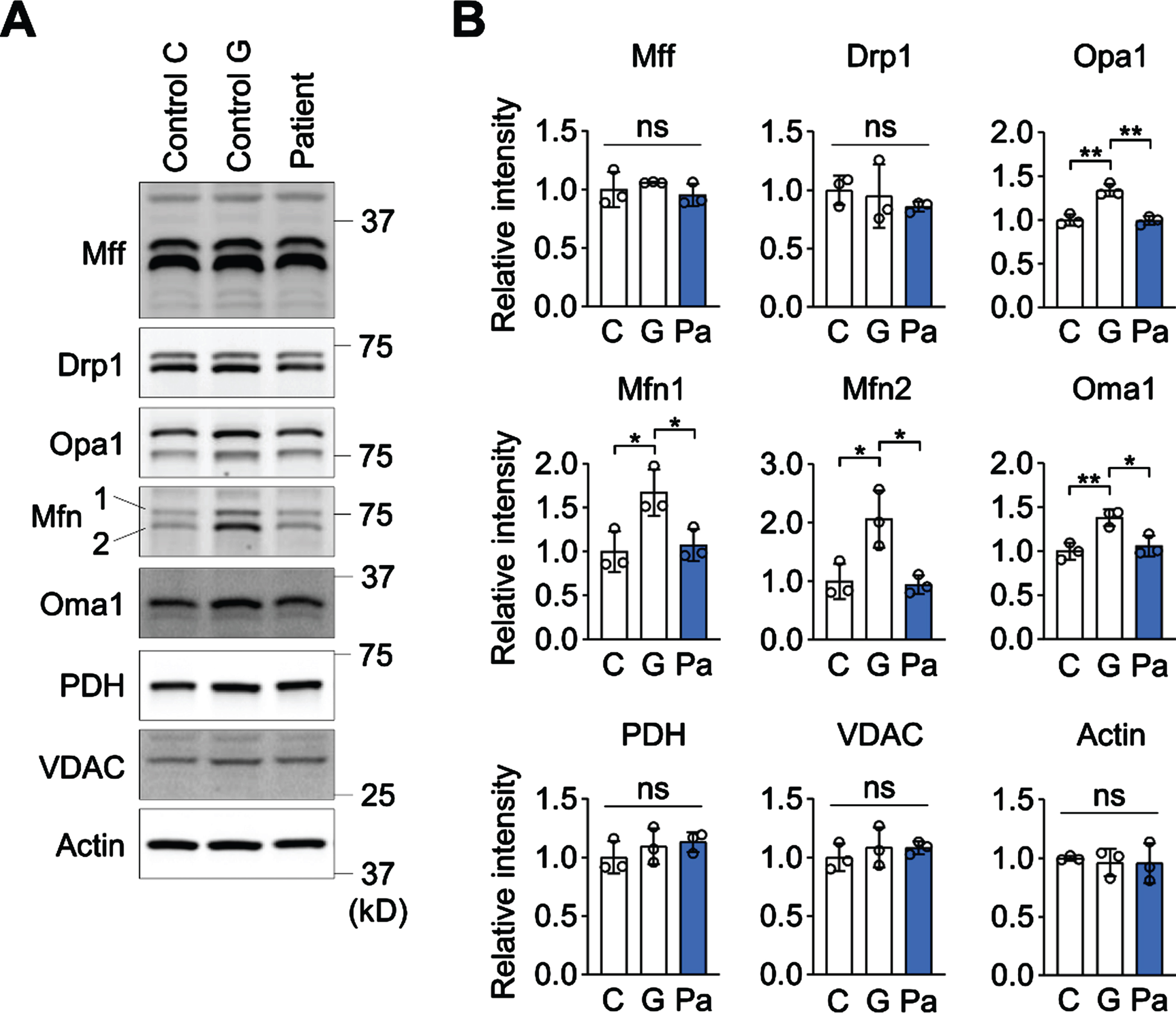
Cells were harvested and lysed in RIPA buffer (9806 S, Cell Signaling Technology) supplemented with cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail (11836170001, Roche) on ice. The lysates were centrifuged at 16,000 g for 10 min at 4°C, and the supernatants were collected. Proteins were separated by SDS-PAGE and transferred onto Immobilon-FL Transfer Membrane (Millipore). The membranes were blocked in PBS-T (PBS containing 0.05% Tween 20) containing 3% BSA at room temperature for 1 h and then incubated with primary antibodies in PBS-T containing 3% BSA at 4°C overnight. The antibodies used were MFF (1:1,000 dilution, gifted from Dr. Alexander M. van der Bliek, UCLA, USA), Drp1 (1:2,000 dilution, 611113; BD Biosciences), Opa1 (1:1,000 dilution, 612607; BD Biosciences), mitofusin 1+2 (1:1,000 dilution, ab57602; Abcam), Oma1 (1:1,000 dilution, sc-515788; Santa Cruz Biotechnology), PDH (1:1,000 dilution, ab110333; Abcam), VDAC (1:1,000 dilution, 4866; Cell Signaling Technology) and actin (1:1,000 dilution, sc-1615; Santa Cruz Biotechnology). The membranes were washed three times in PBS-T, followed by incubation with appropriate secondary antibodies at room temperature for 1 h. After washing the membranes three times in PBS-T, fluorescence signals were detected using a PharosFX Plus Molecular Imager (Bio-Rad). Band intensity was determined using ImageJ software.
RESULTS
Patient evaluation and functional testing of the variant
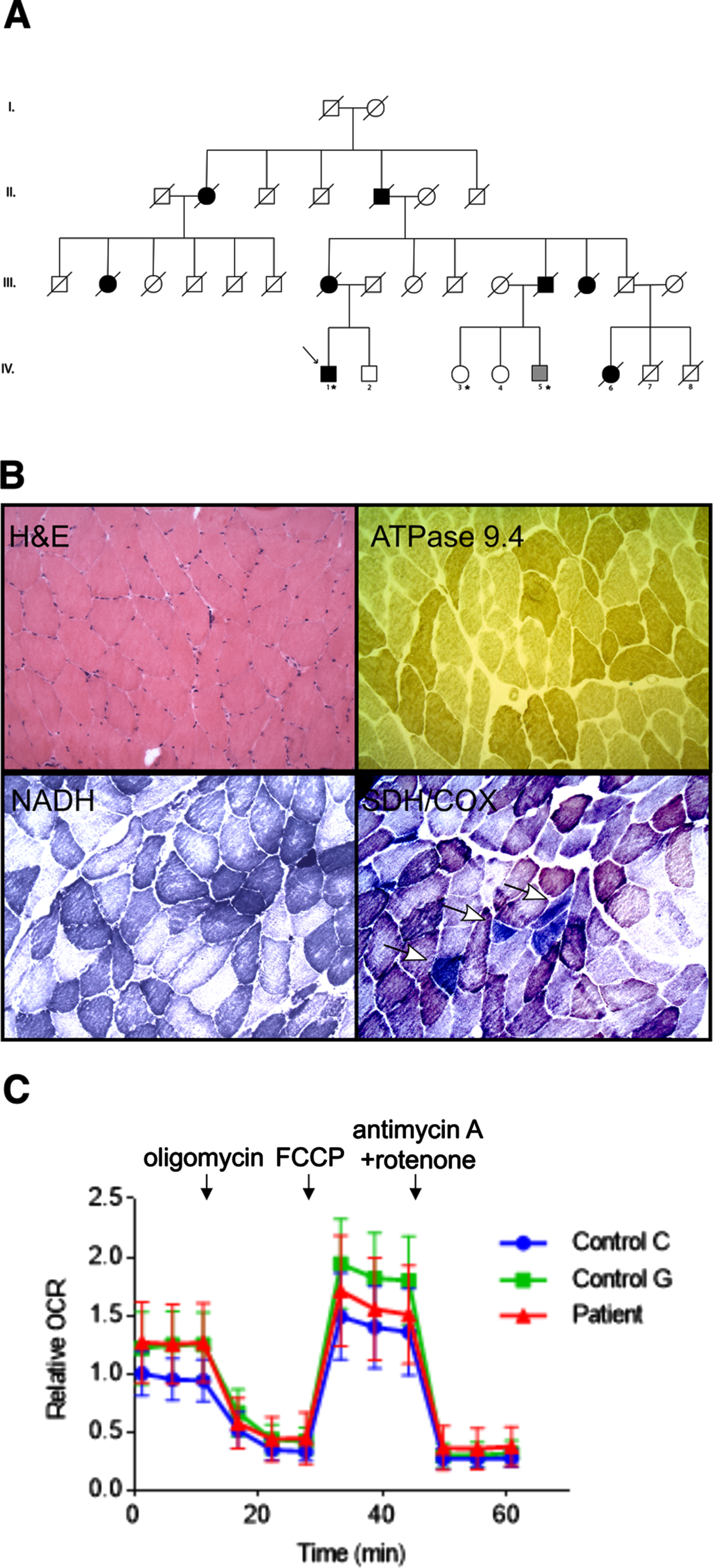
The index patient was initially referred to our institution for evaluation of myasthenia gravis at the age of 76. He had a fifteen-year history of progressive ptosis, and weakness and had been treated for the previous decade with pyridostigmine, prednisone and azathioprine for presumed myasthenia gravis without any relief of his symptoms. There was a family history of other members affected similarly with ptosis, weakness, as well as dysphagia (Fig. 1A). The pattern of inheritance suggested an autosomal dominant manner of inheritance. During our evaluation antibodies against acetylcholine receptor and MuSK were not detected. Needle electromyography (EMG) showed a chronic L5-S1 radiculopathy. Pulmonary Function tests at the age of 80 showed TLC 5.63L (100%), FEV1 1.26 51%), FVC 2.62L (82%). Single fiber EMG of the frontalis muscle showed a mean consecutive difference (MCD) of 20 pairs at 45.69μs, and at least 5 pairs showed jitter and 1 showed blocking. Initial dedicated genetic testing for oculopharyngeal muscular dystrophy (OPMD) revealed no pathogenic expansions in the polyadenylate-binding nuclear protein 1 (PABPN1). Given his lack of response to adequate treatment for myasthenia gravis, and the family history of other affected members, a left biceps muscle biopsy was carried out to evaluate for any underlying genetic myopathy. The biopsy showed increased COX negative myofibers, rounded atrophy and scattered degeneration of predominantly type 1 fibers consistent with a mitochondrial cytopathy (Fig. 1B). Commercial genetic testing, including mitochondrial nuclear gene panel (Supplementary Data), sequencing and deletion analysis did not detect any disease-causing mutations (GeneDx, Rockville, MD, USA). The patient was then referred to a genetic study protocol, and exome sequencing revealed that he was heterozygous for a MFF c.937G>A (p.E313K) variant. This variant has a minor allele frequency (MAF) of 5×10–3 % per data from the gnomAD browser [17]. The exome data was also evaluated for mutations in other common myopathy genes (Supplementary Data). This variant was verified via commercial testing (GeneDx, Rockville, MD). MFF was not included in the initial commercial mitochondrial panel. This variant is predicted to be disease causing by predictive algorithms (mutationtaster). The variant occurs at a highly conserved site in the coiled coil domain of the protein. Its Combined Annotation Dependent Depletion (CADD) raw score was 25.4, PHRED score of 25.4. Given the novelty of this gene we decided to test whether there was a defect in mitochondria and peroxisomes utilizing patient derived fibroblasts. We then decided to see whether the mitochondrial defect was generalized, or whether it was localized only to muscle tissue. To that end, we harvested fibroblasts form the patient and compared oxygen consumption rates to control fibroblasts (Fig. 1C). We did not observe any significant difference in oxygen consumption rates between fibroblasts, which suggested that the mitochondrial defect was expressed preferentially to muscle tissue as evidenced by the biopsy findings. Nonetheless, we reasoned fibroblasts would be helpful in demonstrating the mechanism of action of this particular mutation.
Morphology of mitochondria and peroxisomes is unchanged in E313K muscle tissue and patient derived fibroblasts
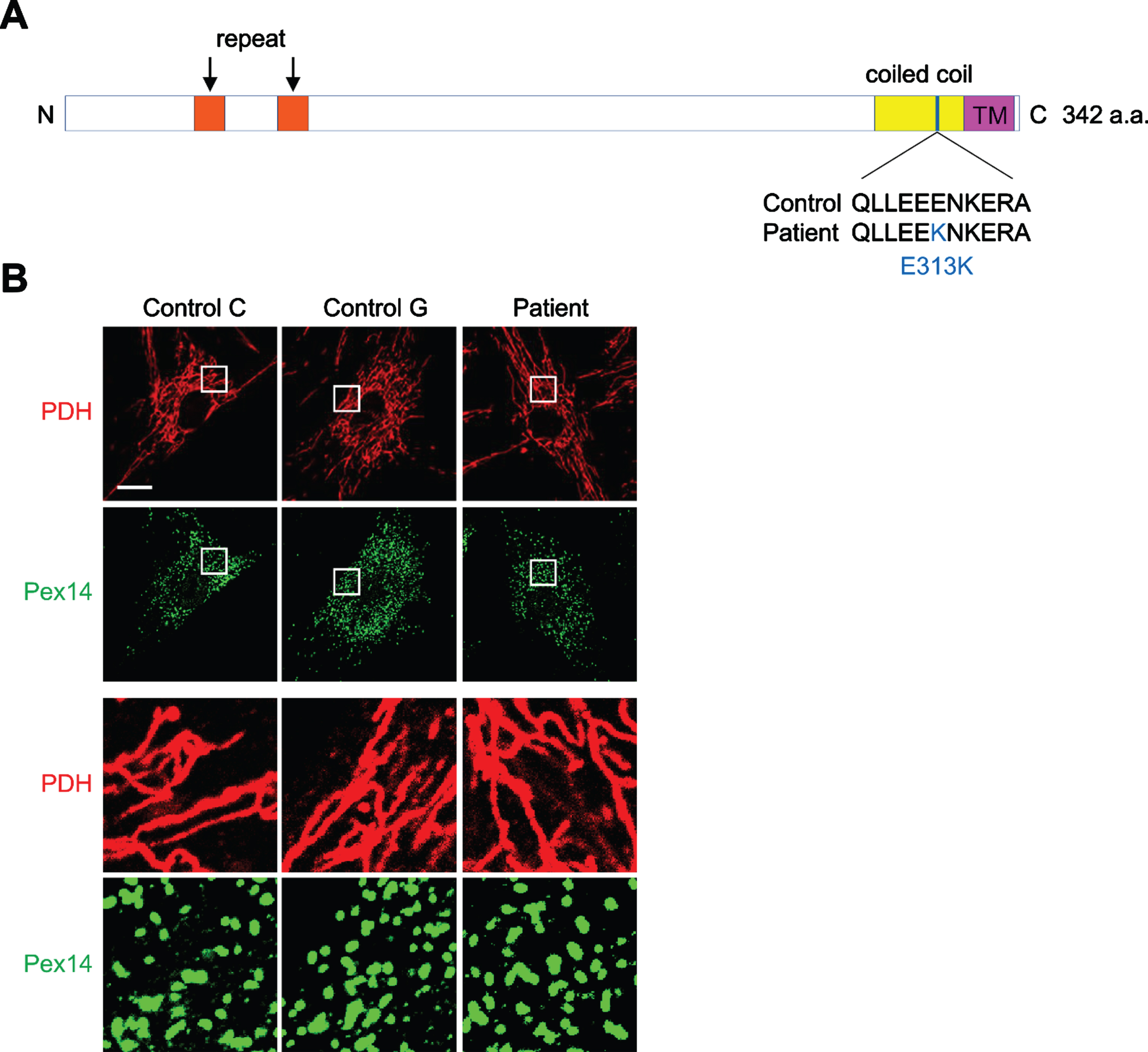
We evaluated the morphology of mitochondria in muscle specimens from the patient and age-matched controls (controls C and G were respectively 59 and 54 y.o. healthy males) using confocal immunofluorescence microscopy with antibodies to a mitochondrial protein, pyruvate dehydrogenase (PDH). We did not observe any obvious differences in the mitochondrial network in the patient muscle (Supplementary Figure 1). We then examined mitochondria and peroxisomes in cultured fibroblasts from the patient as well two other normal controls using confocal immunofluorescence microscopy with antibodies to PDH and a peroxisomal protein, Pex14 (Fig. 2). Again, we saw no morphological difference between the three types of cells.
FCCP-induced mitochondrial division is decreased by the E313K variant in MFF
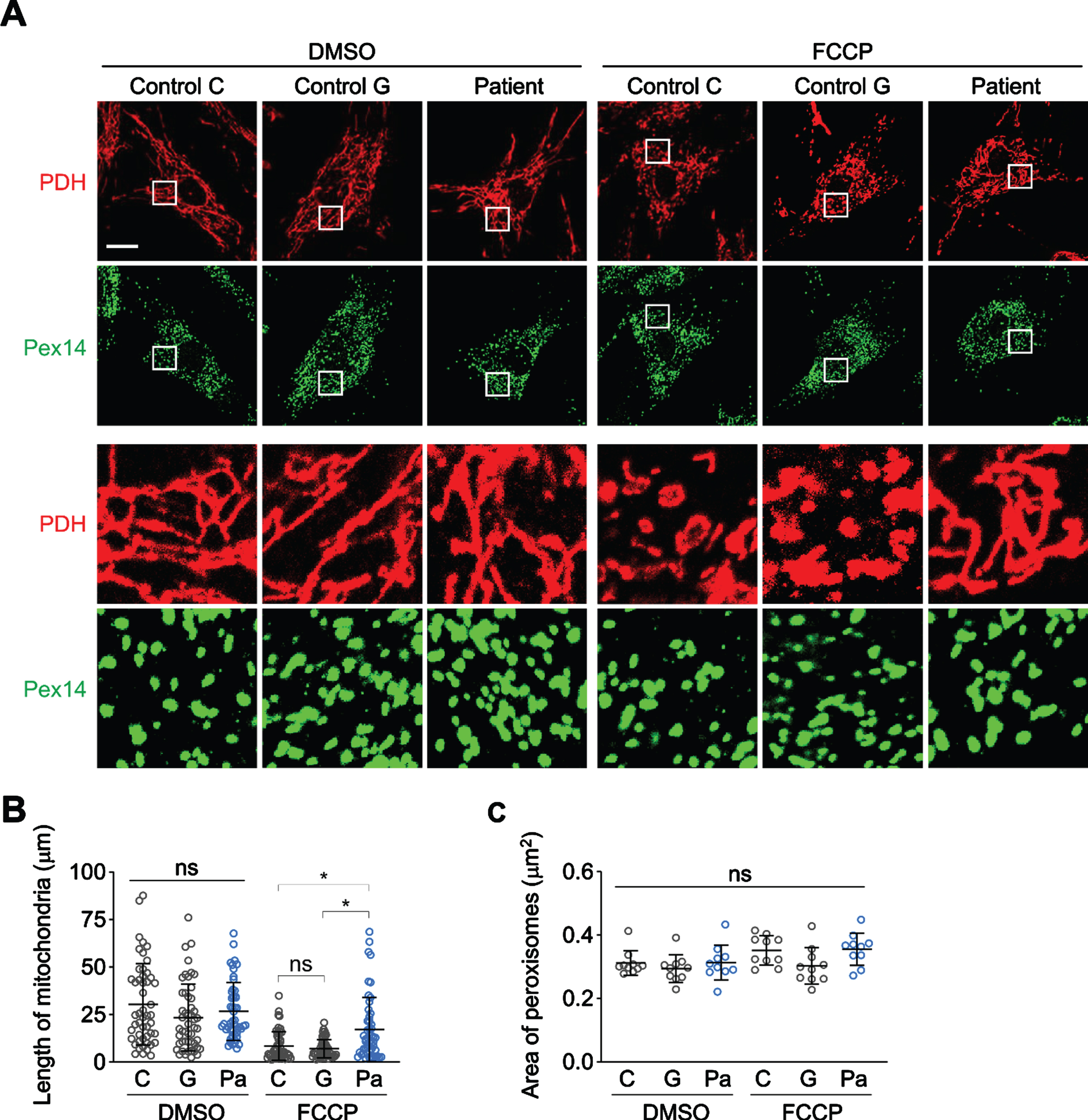
Since MFF plays a major role in mitochondrial division, we assessed the ability of fibroblasts to undergo fragmentation in response to a protonophore, FCCP (carbonyl cyanide-4-[trifluoromethoxy]phenylhydrazone), which dissipates the mitochondrial membrane potential (Fig. 3). We found that in the absence of FCCP there was no statistically significant difference in the length of mitochondria in the patients’ fibroblasts compared to controls. Upon FCCP treatment, mitochondria in both control fibroblasts fragmented. However, in the patient fibroblasts mitochondria became less fragmented. These data suggest a defect in mitochondrial fission under stress in the patient’s fibroblasts (Fig. 1B). As the patient variant in MFF is heterozygous, our results suggest that haplo-insufficiency is the likely mechanism leading to decreased mitochondrial division. When we analyzed peroxisomes, we did not observe changes in their morphology in both control and patient’s fibroblasts.
The localization and expression of MFF are unchanged in E313K fibroblasts
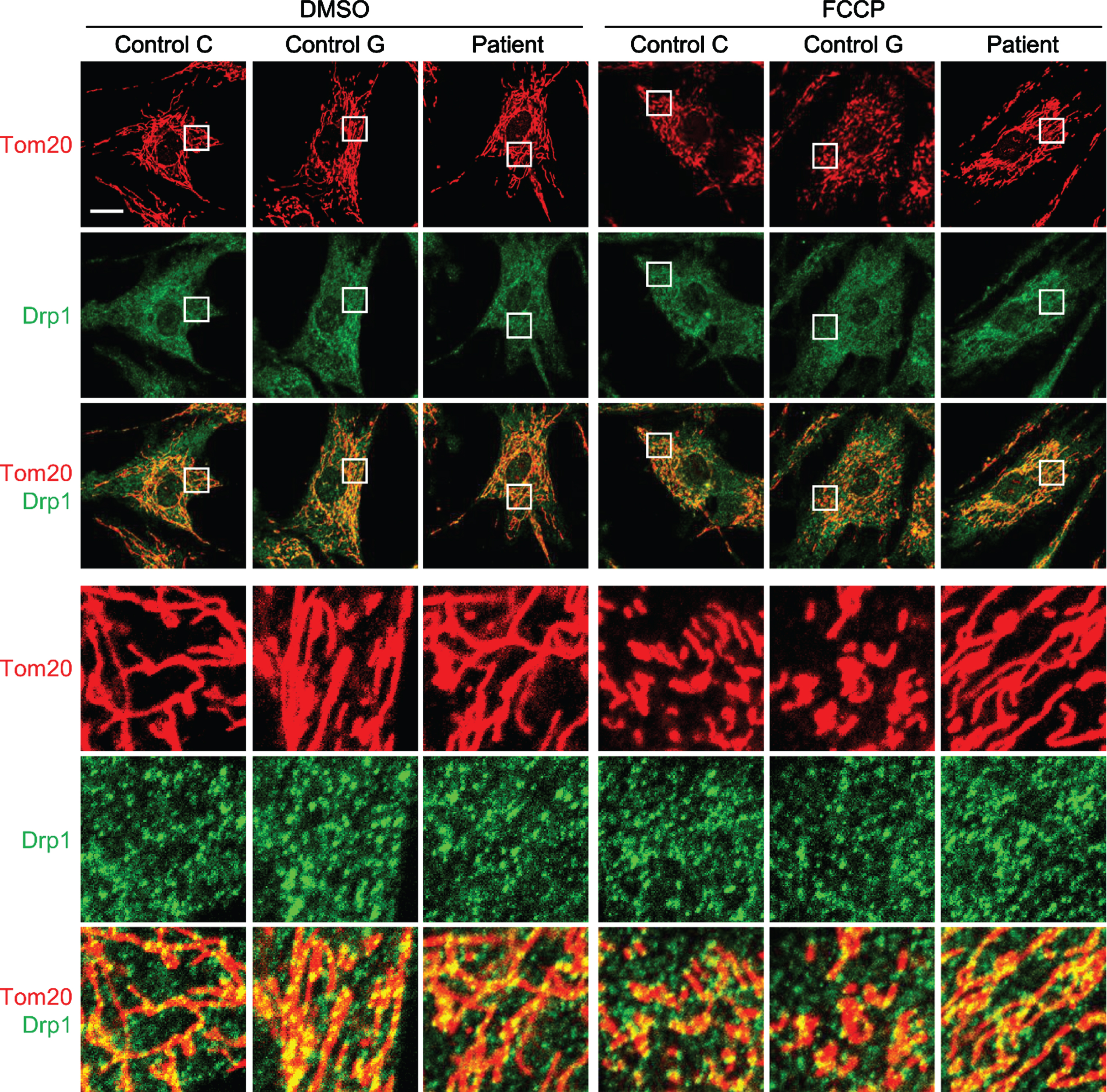
MFF is a receptor protein for Drp1 and recruits Drp1 to sites of mitochondrial division. MFF also activates the GTPase activity of Drp1 and stimulates mitochondrial division. To test whether the E313K variant alters protein to protein interaction and leads to a reduction in the recruitment of Drp1, we performed confocal immunofluorescence microscopy using antibodies to Drp1 and a mitochondrial protein, Tom20 (Fig. 4). We did not observe any alterations in the distribution of Drp1 on mitochondria in the patient fibroblasts in the absence or presence of FCCP (Fig. 3A). We then examined the expression levels of MFF. There were no differences in the expression levels of MFF, Drp1 and Opa1 between the patient fibroblasts and the controls (Fig. 5A and B). The results were in the range of normal controls with respect to the levels of Mfn1 and Oma1, which showed no difference with cells from one control, but were reduced compared to a second.
The E313K variant fails to reconstitute full MFF function
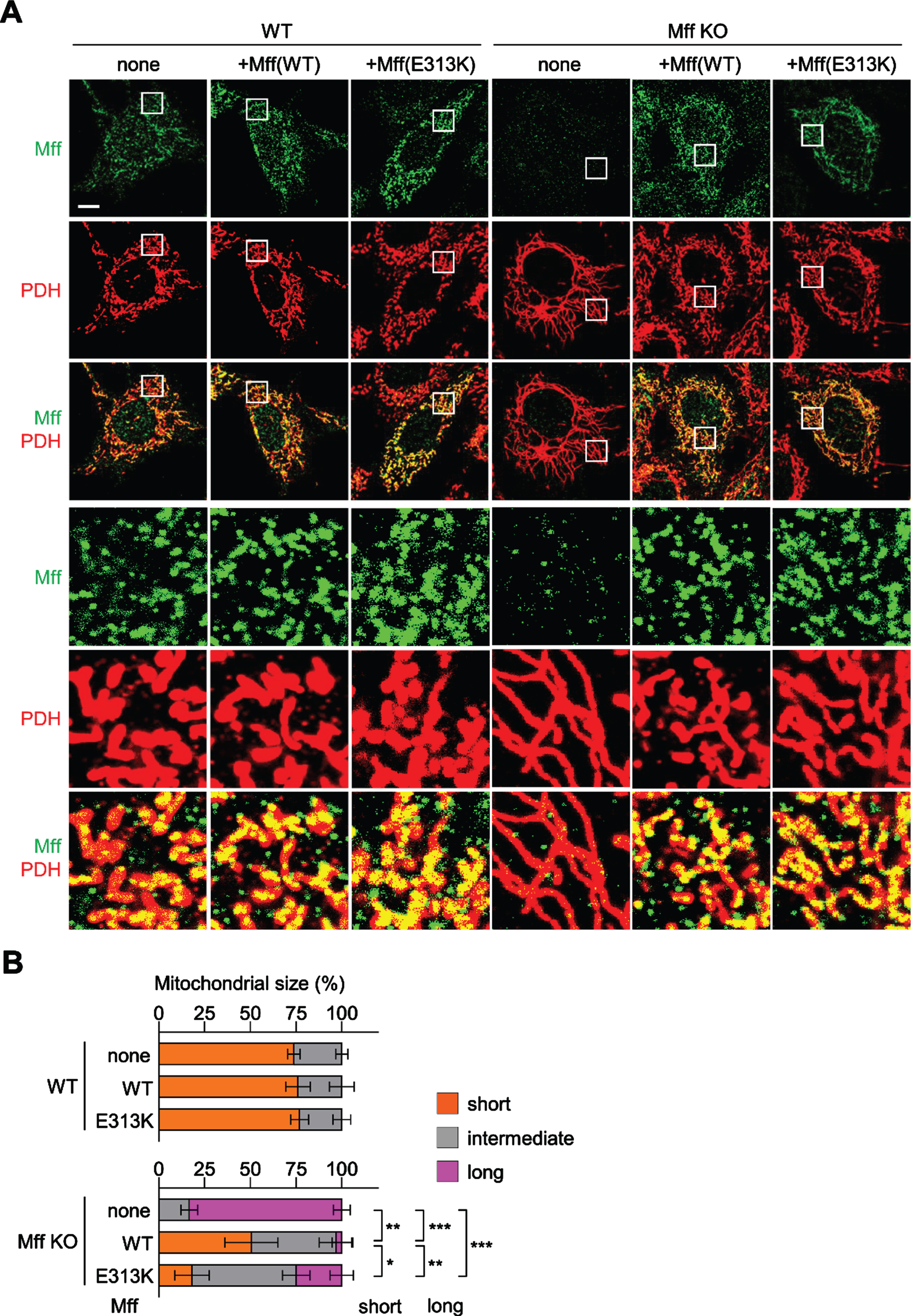
To further evaluate whether the E313K variant is pathogenic and affected the mitochondrial division pathway in fibroblasts, we performed rescue experiments using plasmids which either expressed WT MFF or MFF (E313K) into MFF-KO cells (Fig. 6A and B). In the absence of MFF, mitochondria were elongated compared to WT MEFs. When WT is transfected, 50% of cells contained mainly short mitochondria, while most of the rest showed intermediate length of mitochondria. Expression of the E313K mutant was used to rescue mitochondrial morphology which resulted in less effective rescue of mitochondrial morphology. Compared to expression of WT, there were fewer cells with short mitochondrial tubules and more cells with long mitochondria. These data suggest that MFF (E313K) is not fully functional in mitochondrial division. Expression of the E313K mutant in WT cells, did not lead to abnormal morphology, again indicating that the mechanism of action is haplo-insufficiency, rather than a dominant positive effect.
DISCUSSION
In this manuscript we present a novel variant in MFF which causes a mild myopathy via haplo-insufficiency. The variant is predicted to be pathogenic and presented in our patient with a mild phenotype including ptosis and core weakness. The pattern of inheritance for this variant is autosomal dominant. Given the mechanism of disease, we propose that those affected may not present until later in life with mild phenotypes, such as our patient. This likely contributed to the delayed diagnosis. Recessive mutations in MFF have previously been associated with a severe congenital phenotype [11, 18]. Unfortunately, no complete evaluation of the parents of affected patients is described. Defects in neuromuscular transmission have been noted before in patients with mitochondrial disease [19–22]. In particular, jitter has been observed during SF-EMG, suggesting that remodeling occurs in the neuromuscular junction.
We show using fibroblasts from this patient that heterozygous mutations in this gene lead to a defect in fragmentation in mitochondria. Surprisingly, given the known role of MFF in peroxisomal morphology, we found no defect in peroxisomes. This discrepancy suggests that MFF possibly recruits additional factors needed for mitochondrial fission that are not needed for peroxisomal fission. The defect also appears to be mild, as oxygen consumption rates were not significantly different between controls and the patients’ fibroblasts. The variant is rare, and it is at a conserved site. In vitro rescue studies we carried out show that the variant is unable to compensate for normal mitochondrial morphology, compared to wild type. The importance of identifying variants such as this one is clinically significant in that the phenotypic presentation of the patient was initially suggestive of myasthenia gravis and testing for other genetic mimics was negative. The patient underwent prolonged immuno-suppression, with its concomitant risks for over a decade. We think that additional evaluation of patients with single variants in this gene may further elucidate its function in a mono-allelic background.
Footnotes
ACKNOWLEDGMENTS
This work was made possible, in part, through philanthropic support from Dr. Peter Buck and additional anonymous contributors. CG and AS were supported by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke. We would like to thank the NIH Intramural Sequencing Center (NISC) for whole-exome sequencing and Elizabeth Hartnett, MSW, who contributed as the patient care coordinator for the NIH neurogenetics clinic. MI is supported through NIH grant GM131768. HS is supported through NIH grant GM144103 and also support from the Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation. DC is supported through NIH R35 GM127147.
CONFLICT OF INTEREST
The authors have no conflict of interests to report.