
Abstract
This meeting report summarizes the presentations and discussions held at the summit on ‘Challenges in Gene Therapy’ hosted by the Muscular Dystrophy Association (MDA) in 2024. Topics broadly cover in vitro and in vivo models for understanding of gene therapy related immune responses, strategies to improve safety and efficacy of next-generation gene therapies, and clinical site readiness for gene therapy and post-treatment monitoring. This year's summit also marked the launch of MDA's gene therapy support network – a national resource for patients and clinicians in the MDA care center network aimed at providing sites with the information and resources needed to deliver genetic medicines within our neuromuscular community. A series of recommendations around 8 topic areas including predicting immune responses, long-term follow up of treated individuals, and ethical considerations was derived from content presented at the meeting.
Introduction
The Muscular Dystrophy Association (MDA) held its third annual summit on “Challenges in Gene Therapy using Adeno-Associated Viruses (AAV)”, chaired by Carsten Bönnemann (NINDS, NIH) and Barry Byrne (University of Florida), on January 15–17, 2024 in Tucson, Arizona. Attendees included 41 researchers/clinicians and 34 industry representatives and 16 patient foundation representatives who convened to discuss clinical and non-clinical topics relevant to the translation of AAV gene therapies in the context of neuromuscular disease. Preclinical topics covered this year include gene therapy for exon skipping, translation of next-generation AAV capsids, modeling immune response and durability, and immune predictive assays. Clinical-related topics covered include investigational studies to lower AAV seropositivity, infrastructure and site readiness for gene therapy, and immune response monitoring in treated patients. Challenges and opportunities pertaining to regulatory and economic concerns of gene therapies for rare disease were also explored. The complete list of speakers and their presentation titles are outlined in Table 1.
Listing of meeting presenters and presentation titles.

Gene therapy programs in the clinic
An increasing number of companies are entering gene therapy clinical trials for a range of neuromuscular conditions. For DMD, it is estimated that more than 300 patients to date have received gene therapy as estimated by total enrollment across the five clinical stage DMD programs (Sarepta, Pfizer, Solid Biosciences, RegenxBio, Genethon). Updates for the DMD programs at RegenxBio and Genethon were presented at the meeting, but differences in immune suppression protocols, monitoring period, dose and vector composition across these trials leads to difficulties in making direct comparisons in safety and efficacy.
Since the 2023 meeting, RegenxBio began clinical trials for DMD gene therapy using a novel microdystrophin with the inclusion of a portion of the C-terminal region of the dystrophin gene and the spc5–12 (muscle specific) promoter. RGX-202 is an AAV8 product and has so far been well tolerated in 4–11 year old study participants in a Phase I/II trial. At the time of this meeting, 7 participants had been dosed - three at 1E14 vg/kg and four at 2E14 vg/kg. Biopsies from low dose cohort show robust microdystrophin expression was observed at 12 weeks (western blot and LC-MS), with decrease in serum CK. The trial employs a prophylactic immune suppression regimen which includes eculizumab, prednisolone and sirolimus. Key outcome measures include safety assessments, NSAA, microdystrophin levels in muscle biopsy (at 3 months post-treatment), skeletal and cardiac function assessed by MRI.
Genethon, a non-profit biotech based in France, provided updates on their Phase I-II-III DMD gene therapy program with sites in France and UK. The codon optimized microdystrophin construct was developed by George Dickson and also utilizes the spc5–12 promoter. 1 Based on preclinical data in DMD rats and canines, the minimum effective dose was established at 1E13 vg/kg to 3E13 vg/kg. Accordingly, the first clinical dose cohort was set at 1E13 vg/kg with 2 individuals dosed. Approval was granted for a second dose cohort of 3E13 vg/kg and 3 participants have been dosed to date; with recommendations to move on to pivotal part of the seamless trial with this dose. The design is based on a large natural history/baseline cohort, and it includes a quadruple-blind crossover, with primary read-outs including microdystrophin expression at 8 weeks and changes in NSAA at 52 weeks post-treatment. More centers are opening in other countries in Europe, than in the USA and potentially other non-European countries.
In addition to DMD gene therapy programs, advances in gene therapy products in clinical testing for limb-girdle muscular dystrophy (LGMD) were discussed by Sarepta and Atamyo. These programs are focused on early safety and efficacy as measured by stabilization of disease progression compared to natural history data.
Sarepta's LGMD programs involve delivering genes using self-complementary AAV (scAAV) backbone for more efficient transcription while using the same capsid-promoter combination (AAVrh74-MHCK7). Sarepta's LGMD 2E/R4 program (SRP-9003) enrolled six participants between 4–15 years old for their low (1.85E13 vg/kg) and high (7.41E13 vg/kg) dose cohorts (three for each). Primary outcome focuses on safety, while expression of beta-sarcoglycan will be assessed at 60 days and 2 years post-treatment. Restoration of other proteins in the sarcolgycan complex (alpha, delta and gamma) will also be assessed as endpoints as they have been linked to therapeutic benefit in preclinical models. Sarepta will seek to use the accelerated approval pathway for their LGMD programs by demonstrating restoration of protein expression as a surrogate endpoint and a predictor of clinical benefit. Longer follow-up studies measuring clinical endpoints will subsequently be used to convert accelerated approval to a full approval. Clinical endpoints for Sarepta's LGMD programs include using the North Star Assessment for limb-girdle type muscular dystrophies (NSAD) - a modified version of NSAA used for DMD. NSAD measures items of physical function that are important for patients in daily activities and can be used for clinical efficacy assessment of LGMD gene therapies. Timed function tests such as Timed Up & Go (TUG) and 10/100-min walk tests will also be explored. NSAD is established to be reliable across LGMD subtypes in ambulant and non-ambulant populations. Sarepta's pipeline also includes gene therapies in clinical testing for LGMD 2B/R2 (SRP-6004) and LGMD 2D/R3 (SRP-9004). Programs for LGMD 2C/R5 (SRPT-9005) and LGMD 2L/R12 (SRP-9006) are still at preclinical stage.
Atamyo's LGMD 2I/R9 program (GNT-0006) delivers both the human FKRP gene and a microRNA to silence the transgene in cardiac tissue to prevent tissue-specific toxicity that has been observed in preclinical models. The first clinical cohort of 3 participants received a dose of 9E12 vg/kg, and the second ongoing clinical cohort dose is 2.7E13 vg/kg. Early read-outs so far demonstrate CK reduction, improved changes in forced vital capacity, 10MWT and TUG from baseline. Atamyo's pipeline also includes a gene therapy for LGMD 2C/R5 (GNT-0007) currently in Phase I/II, and LGMD 2A/R1 (GNT-0008) that is in preclinical stage.
A summary of clinical-stage gene therapy programs discussed in our 2024 summit is listed in Table 2.
Summary of gene therapy programs discussed.

Preclinical gene therapy programs
As of October 2024, Citeline reports 90 pre-clinical gene therapy programs in development for neuromuscular disease. Some of these have moved beyond simple gene replacement and are tackling more complicated scenarios such as muti-gene delivery or dominant gain-of-function indications.
Genosera is an early-stage company developing gene therapies aimed at stopping progression of disease and building back lost muscle mass and strength. Genosera's platform uses a dual gene AAV vector that delivers the gene replacement candidate as well as a gene that promotes muscle regeneration. Their LGMD 2I/R9 program is testing delivery of the FKRP and FST (Follistatin) genes in a single vector using the AAVrh74 capsid. Expression of FKRP is controlled by the MHCK7 promoter while expression of FST is controlled by the CMV promoter. The one hour walk test in the FKRP mutant mice demonstrates that dual gene therapy (not FKRP or FST alone) is needed to fully recover ambulation at 7 months at doses of 5E13 vg/kg. Increases in absolute force and myofiber size were more greatly improved with the dual gene vector. These results demonstrate an opportunity to reverse pre-existing disease and rebuild lost muscle strength using gene therapy.
Armatus Bio is an early-stage company focusing on dominant neuromuscular disorders that require knock-down of pathogenic genes. Their primary indication is CMT1A, the most common inherited peripheral neuropathy caused by gene duplication of PMP22. CMT1A is a demyelinating disease that primarily affects Schwann cells. Armatus Bio has designed an artificial microRNA (miR781) to target human PMP22 and reduce its expression to wildtype levels. AAV9 is used to deliver miR781 via lumbar intrathecal injection to transduce Schwann cells. Presymptomatic mice receiving this treatment significantly improved multiple functional outcome measures and nerve conduction velocities, however post-onset treatment show less efficacy in functional behaviors. The most efficacious dose was determined to be 5E11 vg/kg in terms of improving myelination phenotypes in mice, and this dose was further demonstrated to result in successful biodistribution of miR781 in nerve tissue via IT injection. These results provide proof of concept for treating CMT1A using a gene therapy approach.
Exon skipping via AAV
Exon skipping strategies, which are used to put a transcript with an out-of-frame mutation back into frame for a milder phenotype, are applicable to ∼55% of the DMD patient population, with several FDA-approved antisense oligonucleotide (ASO) drugs for DMD targeting exons 51 (Exondys), 53 (Vyondys and Viltepso), and 45 (Amondys).2,3 Despite theoretically restoring near full-length dystrophin, exon-skipping drugs delivered systemically as naked AONs with modified chemical backbones achieve only a modest level of total dystrophin protein (between 0.5–5%). An alternative exon skipping strategy utilizes short antisense RNAs that can exclude exons when fused to a component of a small nuclear ribonucleoprotein (snRNP) complex – the uridine-rich 7 small nuclear RNA (U7 snRNA). U7 snRNAs contain an antisense sequence that it uses to identify pre-messenger RNAs (pre-mRNAs) for their 3′ end processing. Modification of this antisense sequence can turn U7 snRNA into a tool for targeting pre-mRNAs and modifying splicing. 4 Several preclinical studies have demonstrated that AAV vectors can be used to deliver U7 snRNA for exon skipping in DMD models.5–7
RegenxBio presented a U7 sRNA strategy to skip exons 53 or 51 in the hDMDdel52/mdx mice, showing persistence for at least 12 months across multiple skeletal muscles, diaphragm and heart. At a dose of 1E14 vg/kg, the percent of skipped exon peaked at 3 months post-treatment and showed a slight decline over time, potentially due to epigenetic influences that are yet to be understood. Specifically, the delivery of U7 snRNA via AAV at 1E14 vg/kg results in significant dystrophin expression in ∼30% of fibers, while a high dose of 3.5 E14 vg/kg results in significant dystrophin expression in ∼70% of fibers. Delivery of U7 snRNA also leads to a dose-dependent improvement in muscle function test and reduction in creatine kinase. The relationship between percent skipping and dystrophin levels appears to be linear in the heart but not in skeletal muscle or diaphragm for reasons that are unknown. RegenxBio is considering a platform approach in the development of its U7snRNA exon skipping pipeline.
Nationwide Children's Hospital has an ongoing clinical study using the U7 snRNA strategy for skipping of exon 2, targeting patients with the most commonly duplicated exon in DMD. The therapeutic construct contains four copies of the U7 snRNA using self-complementary AAV (scAAV9-U7-ACCA) and is currently being evaluated in a phase 1/2 trial (NTC04240314). Three study participants (Subject 1: 8.9 y.o, Subject 2: 13.7 y.o, Subject 3: 7 months old) have been treated to date at 3E13 vg/kg. About 20% of corrected transcript was detected in subject 1, with less robust correction in subject 2 (<5%). Subject 3 had over 95% corrected transcript which was sustained over a period of 12 months, translating to above 80% of normal dystrophin protein levels and 95–99% positive fibers at 12 months. A decline in the vector genome copy number was further noted in subjects 1 and 2 over 12 months, but not in subject 3. Subject 3, the youngest boy with DMD treated to date with any dystrophin targeted systemic gene therapy, shows normal levels of primarily full-length dystrophin that have persisted until at least 12 months post-treatment. Exon skipping via U7 snRNA may be a pathway for personalized DMD gene therapy and amenable to many classes of mutations, including deep intronic mutations that result in pseudoexon inclusion.
Next-generation technologies: capsids and vector designs
First generation neuromuscular gene therapies, although effective in some indications, will likely be further optimized for potency and safety. Naturally occurring AAVs used in first-generation gene therapies primarily target the liver, requiring very high doses to be effective in disease-affected tissues. These high doses in the range of 1–2E14 vg/kg increase the potential for toxicity and safety issues in patients. Multiple approaches are currently employed to discover and develop AAV capsids with enhanced tropism for tissues of therapeutic interest in neuromuscular diseases and/or decreased tropism for liver. Screening can be performed on construct libraries established through directed evolution or rational design. Directed evolution provides large diversity libraries that explore all possible sequence space, whereas rationally designed AAV selection involves a more targeted approach leveraging scientific understanding of AAV biology to engineer specific modifications. There are pros and cons for each methodology.
Solid Bioscience has pivoted their DMD gene therapy program to utilize a next-generation rationally designed capsid (AAV-SLB101) to enhance skeletal and cardiac tropism. Using AAV9 as the starting capsid and including an RGD containing heptapeptide in the hypervariable region VIII, AAV-SLB101 was selected for its improvements in production yield, binding and uptake into cells, and transgene expression level. In vivo studies using AAV-SLB101 showed a 3–7X increase in biodistribution to skeletal and cardiac muscle, and a 2–3X decrease in liver targeting in preclinical models. Solid is utilizing the newly developed AAV-SLB101 capsid in their IND-approved SGT-003 program, replacing AAV9 as the delivery vehicle for the same genetic cargo as SGT-001. The first cohort of SGT-003 will be dosed at 1E14 vg/kg in an open label study to evaluate safety and microdystrophin expression at day 90.
Kate Therapeutics is exploring next generation capsids across a range of neuromuscular disease programs. Its screening platform involves a machine learning driven directed evolution approach that has resulted in the identification of the MyoAAV and MyoAAV-LD (liver de-targeted) capsid families. The novel MyoAAV capsids consists of variants enabling enhanced systemic muscle and cardiac transduction. MyoAAV capsids contain an RGD domain on the surface and are reported to be potent across mice and non-human primates (NHPs) in vivo and in human myotubes in vitro. One of the first generation MyoAAV capsids was used to design a new gene therapy candidate for XLMTM (KT430) now licensed to Astellas. Newer MyoAAV variants have been evolved to have additional properties such as liver and DRG de-targeting to reduce off-target effects.
Affinia Therapeutics’ approach to identification of potent next-generation capsids consists of AAV backbone modifications and peptide insertions. Screens using an AAV capsid backbone library identified liver de-targeting properties, while screens using a peptide library designed around RGD domain identified myotropic peptides. The resulting “M-series” of capsids show improved muscle and heart tropism with reduced off-target biodistribution in liver and DRGs, properties that translated across mouse and NHPs.
In addition to improvements in capsid technology, there are also opportunities to improve vector design, particularly as it relates to gene replacement strategies for DMD. Jeffrey Chamberlain (University of Washington) presented on a novel strategy to deliver larger and more functional dystrophin transgenes using split intein technology. Split inteins are small polypeptides that carry out a naturally occurring process known as protein trans-splicing, where N- and C-terminal fragments of the protein bind to form an enzyme that catalyzes its own excision and the subsequent ligation of flanking sequences 8 The DMD gene is 2.2MB (13.9 kb cDNA) and current microdystrophin designs are approximately 33% the size of the full-length dystrophin gene. Paired with next generation myotropic capsids, split intein technology enables delivery of larger dystrophin transgenes split across multiple AAVs. 9 A dual vector approach can carry approximately 70% of the dystrophin gene, while a triple vector approach is sufficient to carry the entire dystrophin gene, albeit requiring higher doses due to reduced trans-splicing efficiency. These larger proteins appear to be more stably expressed and result in increased forced production and protection against contraction induced injury. In older dystrophic mice, their expression results in partial reversal of fibrosis and increase in muscle cross-sectional area. Split intein technology can be adapted to deliver other large neuromuscular disease genes that exceed single AAV packaging capacity.
Immune responses in gene therapy
Last year's summit discussions on immunogenicity focused on understanding the immune mechanisms activated by AAV vectors and capsids, identifying mechanisms of complement activation, temporally mapping the kinetics and phases of innate and adaptive immune responses to gene therapy products, and developing immunosuppression strategies. 9 This year, the discussions shifted towards two key areas: (1) using in vitro assays to predict immune responses, and (2) modeling immune responses and durability in animal models to more accurately predict immunogenicity incidence in humans.
Predicting in vitro immune responses
Immunogenicity presents a major challenge to the safe and efficacious development and use of AAV-based gene therapies. Pre-existing neutralizing antibodies to AAV prevent transduction in certain patients, and innate and adaptive host immune responses against the capsid and transgenes, which can occur in any gene therapy recipient, can trigger mild to severe adverse events and immune-mediated clearance of the therapeutic product. During this session, speakers discussed the factors contributing to immune-mediated events observed in previous DMD gene therapy clinical trials and shared their lab's research on using in vitro assays to better understand and predict immune responses.
In DMD gene therapy trials, severe cases of immune-mediated myositis (IMM) occurred that were not predicted by preclinical studies. Helene Haegel (Roche) discussed IMM cases that occurred in patients harboring mutations in the early exons of the DMD gene and treated with AAV-mediated micro-dystrophin gene transfer. The IMM cases was thought to result from a lack of immune tolerance to the N-terminal region of the micro-dystrophin transgene.10,11 In trials by Sarepta-Roche and Genethon, three individuals with deletions involving exons 8 and 9 of the DMD gene developed IMM concomitant with T cell responses to micro-dystrophin. Ex vivo cell-based IFN-γ ELISpot assays revealed two participants with IMM exhibited T cell reactivity to peptides mapping to dystrophin exons 8/9 and exon 8, respectively. In silico epitope mapping based on the HLA alleles of a patient cohort and MHC-associated peptide proteomic assays identified epitope clusters within the micro-dystrophin transgene that might elicit either HLA-Class-I or Class-II mediated T-cell responses. While there was a broad distribution of potentially immunoreactive epitopes found across exons 1–17, the risk of immune responses appeared generally higher for the region encoded by exons 8 and 9 of the DMD gene, highlighting a risk for patients carrying mutations leading to loss of these two exons. In silico epitope mapping based on the patients’ individual HLA-Class-I alleles suggested particularly high risks of HLA-Class-I mediated T cell response to the region encoded by exons 8 and 9 for the three patients who developed IMM. All this points to the risk of IMM for patients with mutations affecting these two exons, now excluded from AAV-based micro-dystrophin gene transfer. However, not all patients with mutations in exons 8 and 9 developed IMM, suggesting that factors such as underlying inflammatory states, immunosuppressive regimens and individual immune variability may contribute to the emergence of individualized immune responses. Thus, these variables complicate the prognostication of immune responses to AAV-based gene therapies and the determination of patient eligibility criteria.
Fraser Wright and Bradley Hamilton (Stanford University) highlighted the need for more robust detection methods for immune responses to guide personalized transient immunosuppression strategies, potentially supported by bespoke immunophenotyping. Achieving this would require a deeper understanding of the intrinsic variables driving immune activation. Fraser identified two key contributing factors: complement cascade activation, particularly C3a and C5a, triggered by high AAV capsid doses, leading to inflammatory cytokine production, and TLR9 innate pathway activation by AAV vector genomes via unmethylated CpG motifs. The following model was presented to describe immunotoxicity: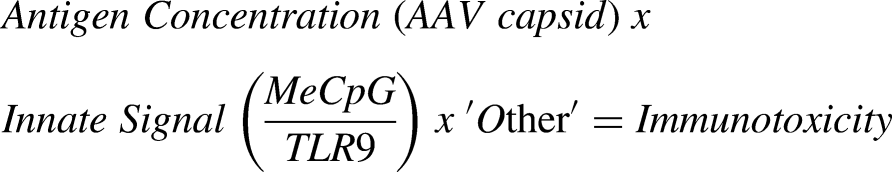
Unlike human DNA, which is largely methylated, rAAV particles are minimally methylated during production, and components like promoters and ITRs, which are of microbial or viral origin, have high intrinsic CpG density. High CpG density can increase TNF and Type 1 interferon levels, and immunotoxicity resulting from downstream mechanisms of TLR9 innate immune receptors can abolish AAV-mediated therapeutic transgene expression in humans. Wright et al., previously published an in-silico prediction model to define the risk factors (NRF3 score) associated with CpGs (methylation status, number of CpGs, stimulatory vs inhibitory CpGs). 12 This model is supported by observations that engineered AAV vectors used in Beqvez and Hemgenix, gene therapies for Hemophilia B, both had <1% CpG density and limited immunotoxicity, while BAX335 (Hemophilia B) had much higher CpG and elicited stronger immune activation. To mitigate CpG-related innate immune activation requires applying a combinatorial approach, including reducing the dose of immunostimulatory CpGs through codon modification, decreasing CpG content in non-ORF components, methylating CpGs during vector production, and increasing cassette efficiency without increasing CpG content. By prospectively immunophenotyping patients and monitoring downstream CpG-related innate immune activation, physicians may be better equipped to create personalized strategies for risk evaluation and mitigation strategies for gene therapy recipients.
However, current methods to detect CpG-mediated immune responses and relevant immune phenotypes is marred by several issues, including: (1) current hPBMC models are poorly responsive to AAV-CpG stimulation, (2) research in animal immune systems is discordant with humans and requires stimulation with overtly immunogenic vectors (3) unreliability of measurements due to donor-dependent variability in INFα1 responses, and (4) traditional measures (e.g., serum transaminase levels) are indirect surrogate markers and assays are generally performed retrospectively.
To address a few of these issues, Fraser Wright and Bradley Hamilton developed an assay using chimeric anti-AAV antibodies to sensitively capture TLR9 activation in response to AAV-CpG stimulation in a human PBMC model. These novel chimeric monoclonal antibodies, composed of a fused Fab region with a human Fc region, distinguish highly immunostimulatory (high CpG content) vectors and those that elicit significant downstream T cell activation. In vitro assays with these antibodies detected immune activation that did not appear through a priori in silico evaluations. The assay also functions as a companion patient screen by capturing donor dependent variations in INFα1 responses. These resources aid in selecting and designing safer AAV genomes that elicit innocuous INFα1 responses based on donor-specific immunophenotyping. Further characterization of vectors using these antibodies demonstrated that reducing ORF TLR9-stimulatory sequences mitigates immune responses more effectively than inhibitory sequences, suggesting the importance of including inhibitory/stimulatory coefficients in in silico models of immune challenges.
There are two main issues for interpreting gene therapy immune phenotypes through current standard laboratory assays (e.g., flow cytometry phenotyping, single cell sequencing, Luminex or flow-based bead assays for cytokine detection). First, blood samples used in these assays are often drawn from the periphery but are used to establish conclusions of local effects and inflammatory status for the individual patient, potentially misleading immunosuppression decisions. Second, assays may not capture direct real-time patient immune status. Further confounding variables, including unclear cell type responses, antigen specificity, and the location of measurements may also obscure data interpretations.
Allison Keeler (UMass Chan Medical School) highlighted the value of ELISpot Assays to investigate antigen-specific T-cell responses, which can be performed in real-time using a peptide library from the AAV capsid or transgene sequence and with incubation of fresh isolated PBMCs. As an example, ELISpot assays were used to evaluate patient immune responses in a clinical trial (GM2) for Tay-Sachs, a lysosomal storage disorder that affects the central nervous system. These patients were delivered an equimolar mix of rAAVrh8HexA and HexB vector dose through direct injection into the thalamus (bilateral) and intrathecally (CSF infusion 75% in Cisterna Magna; 25% at L1–2 level). They were maintained under a strong immune suppression protocol (rituximab ∼6 months, sirolimus ∼60 days, prednisone beginning 5 days prior to vector administration). All the patients showed a positive T-cell response to capsid with responses starting in Week 2, despite adherence to immunosuppressive regimens. Only in 1 patient was there a transient transgene response. After increasing steroid use, liver transaminase (alanine aminotransferase [ALT] and aspartate aminotransferase [AST]) levels and IFN-γ positive ELISPOT responses decreased. Treatment with steroids reduced peripheral capsid-specific T cell numbers; however, some patients did not show ELISPOT IFN-γ responses (capsid-specific T-cell responses) in correlation with transaminase levels. Further phenotyping through flow cytometry analysis for markers of T-cell activation with PBMCs showed induction of capsid-specific Treg cells and not CD4 cells within these patients.
Modeling immune responses and durability in animals
In the past 3 years, 11 severe adverse events associated with AAV products, resulting in deaths, were not adequately predicted by preclinical models. Although current murine models are cost-effective and efficient tools for investigating pathophysiology and treatment efficacy, they fail to fully capture many of these deleterious immune responses in the clinic, partly due to limited conservation of immune system pathways between mice and humans. These innate differences necessitate a careful interpretation of immunogenicity studies conducted in animal models and the development of improved preclinical models for gene therapy research.
Researchers are currently figuring out how to best use mouse models to be predictive of AAV-mediated immune responses. Melissa Spencer (UCLA) discussed how, like humans, the mouse immune system rejects repeated AAV dosing. Administering two doses of AAV, spaced 2–4 weeks apart, induces an immune response in mice against the AAV capsid. In these dual-dosing experiments with AAV9 capsids, the researchers observed classical complement activation within 5 h of the second dose, along with a gradual increase in anti-AAV9 IgG levels starting after the first dose. They also detected significant cross-reactivity when testing sera from AAV9-dosed mice against other AAV capsids, such as AAV8 and MyoAAV1a, but found no antibodies against the transgene itself. In addition to elevated antibody levels and complement activation, myeloid-derived chemokines were induced in a pattern consistent with classical complement activation, peaking 5 h after the second dose, suggesting that myeloid activation is partially dependent on adaptive immunity. To understand how AAV-antibody complexes induce myeloid-derived cytokines release in mice, the team performed single-cell sequencing of myeloid cells, revealing that a specific population of monocytes activates to induce chemokine release after the second dose.
To investigate the relevance of this observation to humans, the lab conducted experiments using human THP1 cells, differentiated into macrophages and exposed to AAV9 in the presence or absence of antibodies, to better understand how AAV is internalized and trafficked. They found that the same chemokines identified in mice were induced in human macrophages, but only in the presence of the anti-AAV9 antibody complexes. These chemokine responses were triggered through Fc receptor (FcR)-mediated uptake, as blocking FcγRs (FcγR1 and FcγR3) attenuated the chemokine responses, highlighting the crucial role of these receptors in mediating antigen-presenting cell activation in response to AAV. These results demonstrate that human elements of the immune response may be replicated in the mouse model and suggest that blocking myeloid cell AAV uptake could be a potential strategy to blunt immune responses.
Other approaches for modeling immune responses to gene therapies and developing sensitive readouts in mice may involve activating the immune system by introducing immunogenic transgene epitopes or priming them through administering components of the immune response.
Armando Villalta (UC Irvine) discussed the immune challenges in mice following in vivo induction with immunogenic dystrophin peptides. The team generated these peptides through an in-silico screen and assessed T-cell activation using an antigen assay. Seven dystrophin peptides were tested; two activated an IFNγ+ CD8 + response, and one triggered an IFNγ+ CD4 + response. Future studies will investigate whether Treg cells can suppress these induced immunogenic responses and promote dystrophin tolerance in a mouse model generated from the crossing of FOXP3DTR with mdx mice. Additionally, the team is actively exploring whether Treg-targeting biologics, including IL-2 complex (IL-2c) and muscle-specific CAR-Tregs can reduce immunogenicity in these induced mouse models. 13
Kanneboyina Nagaraju (Binghamton University) presented data supporting the use of interventions that block phagocytosis/opsonization and endosomal TLR signaling as a pre-gene therapy administration step. 14 In this study, mdx mice were pre-dosed with complement receptor antibodies (CR1, 2, and 3) and TLR 7/8/9 antagonistic oligonucleotides before receiving AAV-µDys. Six of the nine mice developed antibodies against µDys, but only three of the eight mice dosed with CR antibodies and antagonist molecules developed µDys antibodies. Additionally, the treated mice exhibited lower levels of anti-capsid antibodies, higher µDys expression in skeletal muscles, improved grip strength, and reduced body weight gain.
In addition to mouse models, other animals, such as dogs and non-human primates are critical models in translational research to investigate safety, efficacy, and immunogenicity of gene therapies as these models are more physiologically similar to humans.
Hansell Stedman's group compared immunogenicity responses between µDys and the dystrophin-related protein utrophin in the deletional-null German shorthaired pointer dog model. To assess whether central immunological tolerance differs between these gene transfer constructs, the team injected dogs with either 2E12 vg/kg AAV-µDys or AAV-µUtrophin. While focal expression of µDys elicited a robust T cell response, µUtrophin did not trigger any cell-mediated immunogenic response. 15 µUtrophin protected against myonecrosis, while µDys was found in dying cells or those subject to infiltration. The team is currently focused on developing a new gene therapy construct, Nanotrophin, which demonstrates a selective advantage by reducing troponin levels in cardiac muscles compared to conventional µDys.
Non-human primates (NHP) are considered the most suitable preclinical models for investigating immune responses and their kinetics. However, like other animal models, NHP data often exhibit differences compared to human clinical trial results. Juliette Hordeaux (University of Pennsylvania) reported that in NHPs, cellular responses to transgene non-self-products are more intense than responses to the capsid, while humans typically show the opposite response with minimal responses to transgenes (with the exception of µDys). 16 . In a meta-analysis of systemic rAAV administration in 448 NHPs at the Gene Therapy Program at Penn, fewer than 3% experienced serious adverse events warranting an unscheduled necropsy, and among those with a serious event, an increased risk of toxicity emerged when doses exceeded 5 × 1013 GC/kg and within 10 days of dosing. Severe toxicity presentations included transaminitis shortly after dosing, thrombocytopenia, edema, fluid in cavities, bruises, acute liver necrosis, and platelet aggregation in sinusoids. NHP studies attributed the platelet count decrease following high systemic AAV delivery to sequestration in the liver due to endothelial cell activation. Further loss of endothelial barrier integrity caused capillary leakage, worsening edema, fluid build-up, hypoproteinemia, and hemoconcentration. NHP data also suggest that AAV capsid-immune complexes and classical complement activation do not contribute to acute thrombocytopenia, which contrasts with findings in humans.
Collaborative study of Vyvgart to lower anti-capsid antibody in DMD
A pre-requisite of treatment with AAV-based gene therapies is negative (or low) titer of pre-existing anti-AAV antibodies in serum. Seropositivity in patients can arise through natural exposure or previous treatment with an AAV gene therapy product. The exclusion of patients seeking first-time gene therapy due to naturally high titers poses a significance hindrance to treatment in the DMD population. Sarepta's EXPLORE DMD 17 study reported a 14% pre-existing seroprevalence rate (as defined by titers of >1:400) to AAV-rh74 in 101 patients tested, although exact rates across different populations may vary due to geographic or ethnic differences. 18 For this reason, MDA, CureDuchenne and Parent Project Muscular Dystrophy initiated a collaborative effort to support Dr Barry Byrne (University of Florida) in a clinical study to test the use of the approved drug Vyvgart for the reduction of AAV seropositivity in DMD patients. Vyvgart (efgartigimod alfa) is FDA approved for the treatment of myasthenia gravis (MG) in adults. It is an antibody fragment designed to bind with the neonatal Fc receptor (FcRn), inhibiting the recycling of immunoglobulin G (IgG) to effectively lower abnormal acetylcholine receptor antibodies associated with MG. Dr Byrne will investigate whether Vyvgart can be repurposed to lower anti-AAV antibodies. 19 He anticipates that Vyvgart may be helpful for patients who are on the borderline of gene therapy eligibility in terms of seropositivity, but conveys that it would be a bigger challenge to lower seropositivity to the eligible window in those who have previously received gene therapy.
Pre- and post- treatment immune monitoring of gene therapy recipients
Pre- and post- treatment monitoring of gene therapy recipients for immune responses is critical to gain insight into safety and efficacy of gene therapy treatments. Pre-treatment monitoring creates a baseline immunological profile to aid in determining potential adverse events and reduce the efficacy of gene therapy in patients. Post-treatment monitoring enables timely interventions to address any immune-related complications and supports monitoring of efficacy for the administered product.
Francesco Muntoni (University College London) focused on immune responses and vascular pathology in SMA patients with implications for treatment outcomes with Zolgensma. Although in a few instances patients received additional immune-suppression drugs (tacrolimus, mycophenolate) after the gene therapy administration, the incidence of adverse events was more common than previously reported. 20 This might be partly related to an underlying vascular pathology that occurs in SMA (a recent study demonstrated abnormal vascular development in SMA patients, particularly in retinal vasculature, which was corroborated by mouse models). 21 Treatment with SMN-enhancing drugs improved these vascular abnormalities, indicating that the underlying, cell-autonomous vascular pathology is integral to the therapeutic efficacy in SMA and that its correction contributes meaningfully to improved clinical outcomes.
Inflammatory responses before and after treatment with Zolgensma were monitored through advanced mass spectrometry techniques, revealing significant variability in patient immune profiles. Notably, some patients exhibited robust inflammatory responses even before treatment, potentially indicating a predisposition to adverse immune reactions. In several cases, inflammatory markers such as C-reactive protein (CRP) and adiponectin remained elevated despite immunosuppression with high-dose steroids and tacrolimus, indicating variability in the immune response to SMA AAV therapy. Additionally, a novel mass spectrometry method was developed to quantitatively assess anti-AAV (AAV9 specific) antibodies, aiding in identifying patients with pre-existing immune responses. Dr Muntoni concluded by acknowledging the preliminary nature of the research and the ongoing collection of samples for more systematic analysis. The study's findings suggest that SMA patients may have varied immune responses, both before and after treatment, influenced by multiple factors including SMN deficiency and vascular issues.
Diana Bharucha-Goebel (NIH/NINDS and Children's National Hospital) presented data from the Giant Axonal Neuropathy (GAN) clinical trial (NCT02362438) conducted at the NIH. The trial involved intrathecal gene transfer using self-complementary AAV9 vectors (with a synthetic JeT promoter) with immune modulation, including prednisone and rapamycin for all patients at the highest dose, and tacrolimus in addition for participants who are predicted to be CRIM-negative and thus at higher risk for anti-transgene immune responses. Participants, aged 6–14, were symptomatic at the time of gene transfer, with doses ranging from 3.5E13 to 3.5E14 vector genomes. The gene therapy was generally well-tolerated; the most notable adverse event was cerebrospinal fluid (CSF) pleocytosis (lymphocyte predominant), which was clinically silent and responsive to corticosteroids and eventually improved by one year post gene transfer. 22 Significant elevations in anti-AAV9 neutralizing antibodies (NAbs) were observed in both serum and CSF, persisting in serum for up to six years post-treatment (note, CSF collected through 1 year), thereby potentially precluding re-dosing with AAV9 vectors. Cellular immune responses against the AAV9 capsid were detected via interferon-gamma ELISPOT assays from peripheral blood mononuclear cells, which were attenuated by T-cell immunosuppression. No significant cellular immunity against the gigaxonin transgene was observed. Vector genome shedding was detected in blood, saliva, urine, and stool, with a dose-dependent increase and earlier clearance in baseline seropositive individuals. There were no dose-limiting toxicities–although two deaths occurred in the study, these were due to disease progression related complications associated with GAN, not the AAV treatment. The study underscores the challenges of managing humoral and cellular immune responses in AAV-mediated gene therapy, particularly in CRIM-negative patients, highlighting the need for optimized immunomodulatory strategies to enhance long-term therapeutic outcomes and address re-dosing limitations.
Melissa Spencer (UCLA) presented a collaborative project carried out with Armando Villalta (UC Irvine) and Paul Thomas (St Jude) focused on comprehensively characterizing immune responses that arise in DMD patients treated with Pfizer's mini-dystrophin gene therapy. The study involved analyzing peripheral blood mononuclear cells (PBMCs) from pre- and post-dose samples of five patients who received AAV mini-dystrophin, with one patient experiencing a serious adverse event (SAE) of thrombotic microangiopathy (TMA). 23 The PBMCs were stimulated in vitro with AAV peptides and subjected to single-cell RNA sequencing and T-cell receptor (TCR) sequencing without prior enrichment due to the significant cell loss observed during initial enrichment attempts. The sequencing data were analyzed using advanced bioinformatics tools to assess TCR clonality and antigen specificity. 24 The results revealed that all patients developed anti-AAV capsid antibodies post-dosing, with variable titers and temporal patterns. ELISPOT assays demonstrated T-cell responses against the mini-dystrophin transgene in two patients, including the one subject with the SAE, and capsid-specific T-cell responses in most patients. The mini-dystrophin response did not seem to be consequential since none of the subjects in the study experienced myositis. TCR sequencing identified clonal expansions predominantly in CD8+ T effector memory cells in three patients. Notably, in the subject who experienced TMA, the expanded TCRs were largely specific to herpesviruses (EBV and CMV), suggesting that concurrent viral infections may contribute to adverse immune reactions post-AAV therapy. One other subject showed similar TCR expansions to herpesviruses. Additionally, two novel TCR clonotypes were identified that did not match known pathogen-specific TCRs, raising the possibility of AAV-specific T-cell responses. These results emphasize the complexity of immune reactions to AAV-based therapies and underscore the need for further research into immune modulation strategies and the role of coincident infections in adverse events.
Carrie Miceli (UCLA) reported on longitudinal analyses of muscle remodeling in DMD patients, particularly in response to micro-dystrophin gene therapy. Utilizing single-nucleus RNA sequencing (snRNA-seq) on needle biopsy samples collected over a decade, the study examined both myofiber nuclei and resident niche cells within dystrophic muscle tissue. The findings reveal a significant shift in myofiber populations between healthy and DMD samples, characterized by an expansion of immature myonuclei in DMD due to the high demand for regeneration. Notably, patients exhibiting low levels of endogenous dystrophin—arising from spontaneous exon skipping—display myofiber profiles closer to healthy muscle and correlate with attenuated disease severity, as evidenced by clinical scoring and serum creatine kinase levels. DMD myofibers were observed to be immunologically activated, upregulating Toll-like receptor 4 (TLR4) at both the transcriptional and protein levels, as well as expressing HLA (Human leukocyte antigents) Class I molecules and CD80, effectively converting them into antigen-presenting cells. A key discovery is the identification of an expanded subpopulation of fibroblasts in DMD muscle, which exhibits gene expression signatures similar to inflammatory fibroblasts implicated in rheumatoid arthritis. These fibroblasts express complement component C3, Toll-like receptor 4 (TLR4) ligands, and cytokines that influence myogenesis and fibrogenesis, contributing to an inflammatory microenvironment and conditioning the muscle niche through innate immune memory mechanisms. 25 Concurrently, DMD myofibers upregulate TLR4 and antigen presentation molecules, potentially enhancing immune activation. 26 Preliminary data from patients’ post-gene therapy indicate that those with clinical improvement show a shift toward mature myofiber profiles and a reduction in inflammatory fibroblast populations, whereas non-responders maintain inflammatory signatures and exhibit expanded T-cell clonotypes. This suggests that the pro-inflammatory niche and immune responses may impact the efficacy of gene therapies, highlighting the potential benefit of preconditioning strategies to mitigate inflammation prior to treatment.
Gene therapy clinical implementation and readiness
Clinical implementation and readiness for gene therapies involves not only the development and optimization of safe and effective gene delivery systems but also the establishment of multidisciplinary clinical infrastructures capable of comprehensive monitoring for and management of adverse events. Given the complexity of neuromuscular diseases, rigorous clinical protocols should be in place for the safest administration of gene therapy and immediate response to any complications.
Julie Parsons (Children's Hospital Colorado) discussed three clinical cases involving neonates diagnosed with SMA (possessing two or three copies of SMN2 gene) that developed necrotizing enterocolitis (NEC) following administration of Zolgensma within the first three weeks of life. 27 NEC is an uncommon and serious gastrointestinal condition typically seen in preterm infants, making these cases particularly noteworthy due to their occurrence in term infants post-gene therapy. Post-infusion, all infants in these cases developed NEC evidenced by pneumatosis intestinalis on imaging, presenting with symptoms such as emesis, feeding intolerance, vomiting, and hematochezia within 24 h to 19 days after treatment. Laboratory findings included leukocytosis, elevated transaminases, and elevated gamma-glutamyl transferase (GGT) levels; management required nil per os (NPO) status, intravenous antibiotics (e.g., ampicillin and gentamicin), prolonged corticosteroid therapy (methylprednisolone transitioning to prednisolone), and in one case, total parenteral nutrition (TPN) complicated by venous thrombus and anemia. Dr Parsons describes the establishment of a multidisciplinary “gene team” to address the complexities and potential adverse events associated with AAV gene therapy. This team includes specialists in neuromuscular medicine, hepatology, infectious disease, immunology, gastroenterology, pulmonology, cardiology, critical care, nephrology, hematology, rehabilitation, ethics and government affairs and as new adverse events like NEC have emerged, neonatology, to facilitate comprehensive care, close monitoring, and preparedness for emergent situations. Given the potential for unanticipated acute and long-term adverse events such as necrotizing enterocolitis (NEC) and hepatotoxicity, gene therapy must be administered with significant caution. This necessitates extended post-therapy monitoring—including regular assessments of hepatic function, coagulation profiles, and vigilant surveillance for gastrointestinal symptoms. Comprehensive education and communication among multidisciplinary healthcare teams, patients, and families are critical to ensure awareness of potential adverse events and the importance of prompt reporting. Establishing emergency preparedness protocols with access to relevant specialists is essential for timely intervention. Clinicians must stay current with emerging data to adjust monitoring and management strategies. International collaboration and data sharing are imperative to facilitate research, improve patient safety, and inform clinical guidelines. Institutional infrastructures like multidisciplinary “gene teams” are crucial for providing comprehensive care and ensuring patient safety in the evolving landscape of gene therapy.
Diana Bharucha-Goebel (Children's National Hospital and NIH, NINDS) discussed the comprehensive institutional preparedness required for gene therapy implementation, highlighting the necessity of multidisciplinary collaboration prior to FDA approval of novel gene replacement therapy approaches. This includes forming gene therapy working groups involving clinical prescribers, leadership, finance teams, pharmacy, infusion services, and nursing staff. The development of detailed policies, procedures, nursing practice guidelines, consent forms, educational materials, and treatment roadmaps is emphasized to ensure coordinated care. Post-gene transfer monitoring is meticulously planned, guided by prescribing information and sponsor recommendations, with additional check-ins and adverse event reporting mechanisms to mitigate both anticipated and unanticipated risks. Dr. Bharucha-Goebel presented an informative case involving an 8-month-old male infant with symptomatic Type 1 SMA who developed significant hypertension following administration of Zolgensma. The patient had a baseline anti-AAV9 antibody titer of 1:25, which, although within the allowable range for dosing (<1:50), prompted heightened vigilance. On Day 4 post gene transfer, the patient exhibited vomiting, poor oral intake, decreased urine output, and was found to have hypertension at the 99th percentile for his age, despite being clinically well-appearing. Laboratory evaluations revealed mild transient transaminitis, troponin elevation, a decrease in complement component C3, and an increase in C5b-9 levels, raising concerns for possible complement-mediated thrombotic microangiopathy (TMA). 28 Ultimately the workup for TMA was negative, the urine output improved with fluids, and the hypertension was managed with antihypertensive therapy. The patient later tolerated gradual corticosteroid weaning at 14 weeks post gene transfer and eventually the anti-hypertensive therapy was successfully stopped. The case underscores the importance of close monitoring for hypertension and renal function post-gene therapy, the potential for steroid-induced hypertension in infants, the role of complement activation, and the need to consider pre-existing anti-AAV9 antibodies as a risk factor for adverse events.
John Day (Stanford University) addressed the challenges of predicting safety and efficacy responses to gene therapy in human clinical trials, emphasizing the significant genetic diversity and variability among patients compared to preclinical mouse studies. Dr. Day underscored the challenges in identifying consistent patterns due to inconsistent data across patients and highlights the necessity of assembling standardized, comparable data from multiple sites to achieve statistically meaningful conclusions. Lessons learned from Zolgensma therapy include observation of adverse events such as severe nausea, vomiting, thrombocytopenia, biphasic hepatocellular injury, and notably, significant elevations in troponin levels indicative of cardiotoxicity. While corticosteroids have been employed to mitigate these effects, their efficacy remains uncertain due to the associative rather than causative evidence. Detailed clinical cases involving DMD patients illustrate the multifaceted and severe adverse reactions in patients post-AAV therapy, including cardiac and hepatic complications, and the limitations in current monitoring and management strategies. Dr. Day advocates for the development of standardized templates, surveillance committees, and protocols to enhance data collection and enable cross-site collaborations. The critical need for financial support is emphasized, suggesting that revenue generated from high-cost treatments be allocated to support these initiatives and that patient organizations play a pivotal role in leveraging resources. Additionally, he stresses the importance of obtaining autopsy data to elucidate the pathophysiology of adverse events, noting the paucity of such data despite multiple fatalities, which hampers the ability to fully understand and address the safety concerns associated with AAV gene therapies.
Michael Storey (Nationwide Children's Hospital) discussed the complex financial and administrative challenges in securing insurance authorization for gene therapies, specifically highlighting the obstacles in obtaining timely approvals for treatments like Zolgensma for infants diagnosed with SMA. The necessity of meticulous preparation in crafting letters of medical necessity that are concise, patient-specific, and directly aligned with the insurer's medical policies are emphasized. This approach aims to simplify the approval process by directly addressing the criteria outlined by insurers, thereby reducing the likelihood of denials based on misinterpretations or lack of information. The insurance denials are categorized into clinical and administrative types, noting issues such as patients not meeting plan criteria, insurers misinterpreting clinical data or their own policies, and the significant problem of benefit exclusions in employer self-funded plans. Dr. Storey explains that some employers inadvertently exclude coverage for all gene therapies, leading to substantial financial liabilities when treatments are needed, as stop-loss insurance does not cover excluded benefits. This situation can impose million-dollar expenses on employers for a single treatment. Dr. Storey suggested that the community should advocate for proactive measures, urging employees to review their insurance plan documents to ensure gene therapies are covered and to address exclusions before they affect patient care. Additionally, the operational challenges faced by healthcare institutions, including delays in reimbursement, cash flow issues, and the necessity for coordinated efforts between pharmacy, finance, and clinical teams are discussed. He pointed out that Nationwide Children's Hospital institution has established a Gene Therapy Center of Excellence to navigate these challenges, aiming to optimize resource allocation and streamline processes for emerging gene therapies.
Regulatory and economic barriers to gene therapy development in the us and Europe
Paul Melmeyer (MDA) discussed the regulatory and economic barriers facing gene therapies in the United States, using Elevidys as a case example. As the first gene therapy approved via the FDA's accelerated approval pathway for a neuromuscular disease, Elevidys introduced unique challenges for patient access. Insurers have often cited the use of the accelerated approval pathway and the limited efficacy data available at the time of approval as reasons to deny coverage. Although there are well-established clinical outcome measures in DMD that could potentially support value-based payment (VBP) models, the difficulty in demonstrating clear efficacy for Elevidys using these measures — combined with the drug's high cost — complicates the implementation of such agreements. Despite these impediments, some patients have successfully gained access to the therapy.
In the United States, regulatory and policy frameworks have supported the development and approval of gene therapies, particularly for rare and genetic diseases. The FDA has enabled access to accelerated approval pathways and has demonstrated regulatory flexibility, especially in the context of high unmet medical need—an approach that has made the U.S. a leader in the number of approved gene therapies globally. Programs such as the Orphan Drug Designation (ODD) continue to provide important incentives for sponsors, including 7-year market exclusivity, user fee waivers, and clinical trial tax credits. The Rare Pediatric Disease Priority Review Voucher program has also served as a strong incentive to advance gene therapy development for pediatric conditions, though the program is currently set to expire in September 2024 and its reauthorization remains uncertain. To further support rare disease development, the FDA has introduced initiatives such as the START (Support for clinical Trials Advancing Rare disease Therapeutics) pilot program, and continues to strengthen its internal infrastructure and review capabilities to accommodate the growing pipeline of gene therapy products. Still, further policy development and enactment may be warranted to address the commercialization challenges associated with a prospective market size of fewer than 100 patients, as is often the case with ultra-rare genetic neuromuscular diseases for which gene therapies may be possible.
In Europe, Gérald Perret (Genethon) provided an overview of the regulatory landscape for gene therapies. At the time of the meeting, Elevidys was not expected to receive marketing authorization from the European Medicines Agency (EMA), largely due to concerns around the limited efficacy data available from placebo-controlled trials. While the EMA has since validated and initiated review of the marketing authorization application for Elevidys in 3–7-year-old Duchenne muscular dystrophy (DMD) patients (as of June 2024), regulatory approval remains uncertain. At the time of writing, the EMA has placed three clinical trials of Sarepta's Elevidys on hold in Europe pending an investigation into two deaths linked to liver failure. Unlike the U.S., Europe operates under a centralized regulatory framework for gene therapies, with nearly all advanced therapies—including gene therapies—requiring approval via the EMA's centralized Marketing Authorization Application (MAA) pathway. Although individual European countries have their own health systems and pricing authorities, national regulatory approvals outside the centralized EMA process are rare and generally limited to non-EU states or exceptional circumstances (e.g., post-Brexit UK). Following EMA approval, gene therapies must undergo additional review by national Health Technology Assessment (HTA) bodies, which evaluate clinical and economic value to determine pricing and reimbursement. A major hurdle for gene therapies in Europe is the difficulty of justifying their high upfront costs within traditional HTA frameworks based on quality-adjusted life years (QALYs) and cost-effectiveness thresholds. This stands in contrast to the U.S., where pricing is less constrained by centralized cost-benefit evaluations and more driven by market dynamics.
Europe has introduced programs to streamline approvals across European countries, including introducing the Clinical Trial Information System (CTIS) on January 2023 for sponsors to apply for authorization to conduct clinical trials across up to 30 EEA countries through a single application, and the Joint Clinical Assessment (JCA) pilot program in June 2023 for clinical assessment of health technologies (medical devices) and to coordinate evaluations across states. The EMA has 2 programs that are similar to the US FDA: 1) Orphan Drug Designation for rare diseases, which provides fee reductions up to 100%, protocol assistance, scientific guidance, 10-year market exclusivity vs similar products, and 2) Priority Medicines (Prime) designation, which provides improved scientific guidance through an EMA dedicated TEAM, total fee exemption for scientific guidance, and expedited scientific follow-up, sharing many similarities to the US FDA START program. Individual country-specific programs also exist for supporting access to therapies. For example, in France, early access can be requested by the sponsor and compassionate access can be requested by prescribers (e.g., physicians) for their patient and authorized by the French regulatory authority for one-year with renewals available on a per-year basis.
Despite regulatory advancements, several challenges persist for the broader adoption and reimbursement of gene therapies in Europe. Health Technology Assessment (HTA) processes remain a key area of contention due to the challenges many gene therapy programs face in justifying high costs in terms of improved patient quality of life. Market access is further complicated by Europe's fragmented healthcare landscape, which comprises multiple countries, diverse payer systems, and differing national priorities. Additionally, pricing constraints can discourage drug developers from seeking regulatory approval in many countries, significantly limiting patient access. Although HTA bodies typically conduct evaluations after EMA approval, earlier dialogue between sponsors, the EMA, and HTA agencies—ideally during late-stage development (e.g., Phase II or early Phase III)—could help align expectations regarding evidence generation, clinical endpoints, and value demonstration. While the full comparative effectiveness and cost-benefit of a therapy are typically assessed post-approval, early engagement may enable developers to design trials that generate data relevant to HTA requirements, potentially reducing delays in national reimbursement decisions. It is important to distinguish between the EMA's centralized regulatory review (typically completed within 12–15 months) and subsequent country-specific HTA evaluations, which can vary considerably in scope and timing. Indeed, the HTA process is marked by substantial heterogeneity across Europe in terms of methodology, timelines, and thresholds for cost-effectiveness—factors that may affect access to gene therapies regardless of EMA approval. This variability has been recently highlighted in a systematic review documenting the diverse national approaches to HTA decision-making in the EU. 29
MDA gene therapy support network
Nora Capocci (MDA) reported on MDA's initiatives to provide resources for the neuromuscular disease community and clinical experts, with the goal of ensuring safe and equitable access to gene therapies. In response to the imminent accelerated approval of AAV gene therapies—such as Elevidys for Duchenne Muscular Dystrophy—and the development of approximately 90 AAV therapies for neuromuscular diseases in the pipeline, the MDA identified a critical need to support both clinical practitioners and the patient community. The Gene Therapy Support Network (GTxSN) was established to create a centralized hub for resources aimed at ensuring safe, efficient, and equitable access to these novel treatments. This initiative focuses on establishing a clinical operational readiness network within the 150 center plus MDA Care Center Network, optimizing safety and long-term outcomes through resource provision, and developing educational materials to inform patient and caregiver decision-making.
The network is structured around three synergistic pillars: bolstering MDA Care Center and community support, facilitating best-practice sharing among clinical experts, and enhancing community education. A key component is the Care Center Information Hub, designed to equip MDA clinical sites with standardized protocols, resource checklists, and guidelines for adverse event monitoring across four critical areas: (1) institutional preparedness; (2) clinical coordination and capacity management; (3) in-hospital coordination; and (4) family communication. Concurrently, the MDA Gene Team provides dedicated support to patients and caregivers through insurance navigation assistance, virtual consultations, and multilingual educational resources, including workshops and myth-busting documents. This comprehensive approach aims to harmonize clinical practices, address gaps in care delivery, and foster informed decision-making within the neuromuscular disease community, ultimately enhancing the readiness and capability of care centers to implement gene therapies safely and effectively.
Concluding remarks
The 2024 Muscular Dystrophy Association Summit on “Challenges in Gene Therapy” brought together a diverse group of researchers, clinicians, industry leaders, and patient foundation representatives to address the multifaceted challenges facing the development and implementation of gene therapies for neuromuscular diseases. The summit highlighted significant advancements in both clinical and preclinical gene therapy programs, including those targeting DMD, LGMD, and other neuromuscular disorders.
Clinical programs showcased promising results with novel gene therapy constructs and delivery methods, emphasizing the importance of standardized protocols and the need for robust immune suppression strategies to mitigate adverse immune responses. Preclinical studies introduced innovative approaches such as bicistronic vectors for muscle regeneration and artificial microRNAs for gene silencing, expanding the therapeutic possibilities beyond gene replacement.
A critical focus was placed on understanding and managing immune responses to AAV-based gene therapies. The discussions underscored the limitations of current animal models in predicting human immunogenicity and the necessity for developing more predictive in vitro assays and humanized models. The summit also highlighted the complexities of pre- and post-treatment monitoring, emphasizing the need for comprehensive immune profiling to enhance patient safety and therapeutic efficacy.
The launch of the MDA Gene Therapy Support Network represents a significant step toward addressing the infrastructural and educational gaps in gene therapy implementation. By providing resources and fostering collaboration among care centers, the network aims to ensure equitable access and optimize patient outcomes.
Regulatory and economic barriers were acknowledged as significant hurdles, with discussions emphasizing the need for proactive engagement with regulatory bodies and the development of strategies to navigate the complex landscape of gene therapy approval and reimbursement, both in the United States and Europe.
Overall, the summit fostered a collaborative environment that catalyzed discussions on overcoming the scientific, clinical, and logistical challenges in bringing gene therapies from bench to bedside. The collective insights and shared experiences of the participants have laid the groundwork for actionable strategies to advance the field. The actionable meeting recommendations for implementation by key stakeholders in the field of neuromuscular diseases are summarized as follows:
Footnotes
Acknowledgements
The Muscular Dystrophy Association would like to acknowledge Sarepta Therapeutics, AskBio, Pfizer, Armatus Bio, Kate Therapeutics, Astellas and RegenxBio for contributing to the meeting sponsorship.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.