
Abstract
Satellite cells, the resident muscle stem cells, are essential for skeletal muscle post-natal growth and regeneration. Dysfunction in these cells contributes to a group of muscle disorders known as satellite cell-opathies, which can be categorized into primary and secondary forms. Primary satellite cell-opathies stem from intrinsic defects within satellite cells, including genetic mutations that impair their survival, self-renewal, proliferation, or differentiation. Alternatively, secondary satellite cell-opathies result from pathological conditions affecting both the satellite cells and the muscle fibers. This review explores the pathophysiology of satellite cell-opathies, highlighting key molecular mechanisms underlying their dysfunction. Additionally, we discuss emerging therapeutic approaches, including gene therapy, pharmacological interventions, and cell-based therapies, which aim to restore satellite cell function and promote muscle regeneration. A deeper comprehension of these mechanisms and satellite cell–targeted strategies is essential to drive the development of innovative therapies for this emerging class of muscle disorders.
Keywords
Overview of satellite cells and myogenesis
Satellite cells were originally reported by Mauro in 1961 using electron microscopy. 1 He correctly hypothesized that satellite cells are remnants of embryonic development that failed to fuse with other myoblasts, remaining dormant yet poised to recapitulate embryonic myogenesis when the main multinucleated muscle fiber is damaged. This myogenesis process is tightly regulated and occurs throughout life: from embryonic development of skeletal muscle, through postnatal muscle growth, and into adulthood during regeneration following injury. This review highlights the distinct cellular and molecular mechanisms governing satellite cells in the different stages of myogenesis, as well as the impact of genetic variants on satellite cell function in myopathies. In particular, we distinguish primary and secondary cell-opathies based on the classification originally described by the group of Peter Zammit2,3 and discuss emerging approaches to target these defective cells for the treatment of satellite cell-opathies.
Embryonic origin of satellite cells and developmental myogenesis
In vertebrates, embryonic development is characterized by the formation of three different germ layers: the ectoderm, endoderm and the mesoderm. From the mesoderm, cells aggregate to form the somite structure, which give rise to the skeletal muscles of the body and limbs, as well as dorsal dermis, cartilage, tendons, vertebrae, and ribs. The dorsal portion of the somites, named dermomyotome, contains the progenitor cells that delaminate and differentiate to form the underlying myotome. 4 The majority of progenitor cells in the myotome are actively proliferating and co-express the transcription factors Paired box 3 (Pax3) and Pax7. 5 These paralogous factors have overlapping functions, yet Pax3 is indispensable during embryonic myogenesis for somite patterning and the formation of the dermomyotome lips that will give rise to migratory myogenic cells destined for limb muscles, whereas Pax7 is critical postnatally for the specification and survival of satellite cells.6,7 In response to signaling molecules such as Wnt (Wingless), BMP (Bone Morphogenetic Protein), and Shh (Sonic Hedgehog) released from surrounding structures these progenitor cells will turn on the expression of helix-loop-helix (bHLH) myogenic regulatory factors (MRFs) Myf5 (Myogenic factor 5) and MyoD (Myoblast determination protein 1) to become myoblasts. 8 MyoD and Myf5 exhibit overlapping functions in myogenic determination. In mice, when MyoD is deleted, mice exhibit slightly delayed but normal formation of skeletal muscle due to compensation by Myf5 overexpression. 9 Similarly, mice lacking Myf5 experience mild delay but do not display skeletal muscle abnormalities during development due to MyoD compensation, although they die shortly after birth due to breathing complications caused by rib deformities. 10 A more severe phenotype has been observed when both the determination factors MyoD and Myf5 are inactivated simultaneously, which leads to absence of myoblasts and skeletal muscle, and perinatal death. 11 Importantly, because Myf5 and another MRF, Mrf4 (also known as Myf6), are located in close proximity within the same genomic locus and share common regulatory elements, the observed phenotype can vary depending on the extent to which Mrf4 expression is secondarily affected by the genetic targeting of Myf5. It was shown that muscle differentiation can be initiated if the expression of Mrf4 is maintained in MyoD/Myf5-double knockout, highlighting Mrf4 as a determination gene. 12 MyoD promotes cell cycle exit through regulation of cyclin/CDK activity, and participate in chromatin remodelling to activate the transcription of silenced genes such as Myogenin (Myog) that plays a key role in myoblasts differentiation. 13 Knockout of Myog in mice results in the presence of myoblasts that are not able to form functional multinucleated fibers leading to muscle deficiency and perinatal death. 14 The fusion of myocytes marks the final step of differentiation leading to the formation of myofibers. This fusogenic process is regulated by two key proteins: Myomaker and Myomixer (also known as Minion or Myomerger).15–17 The former is required for initiating hemifusion, while the latter mediates the subsequent formation of fusion pores.18,19 The Myomarker (Mymk)-null embryos exhibit severe muscle deficiencies and absence of multinucleated fibers at birth. 19 Similarly, mice knockout for Myomixer (Mymx) exhibit severe myoblast fusion defects during development and minimal muscle formation. 15
Developmental myogenesis happens in successive waves. After the initial myotome formation in the somite around E8.5-E9.5 in mice, a portion of the myoblasts fuse to form the primary muscle fibers (embryonic fibers) around E10.5-E12.5, while remaining myoblasts continue to progress through myogenesis to form secondary muscle fibers (fetal fibers) between E14.5-E17.5. 20 Pioneer study in rats showed that while myoblasts are uniformly distributed throughout the developing muscle, secondary myotubes are formed exclusively within the innervation zone of primary myotubes. 21 These secondary fibers surround the embryonic fibers, forming a ring-like configuration. Newly formed myofibers will start expressing contractile proteins, such as Myosin Heavy Chain (MyHC), particularly the embryonic (MYH3) and neonatal (MYH8) isoforms, before expressing the mature isoforms (MYH1, MYH2, MYH4, MYH7). Deletion of Myh3 expression in mice resulted in lower body weight, higher proportion of small fibers, and scoliosis. 22 It also induced a non-cell-autonomous effect resulting in the depletion of the myogenic cell pool. An increased expression of other MyHC isoforms (Myh1, Myh2, Myh8) was also observed to compensate for the loss of Myh3.
Importantly, by E16.5, a subset of progenitors locates between the sarcolemma and the forming basal lamina, establishing the satellite cell niche, which will host the stem cells responsible for postnatal muscle growth and regeneration. 23
Postnatal growth
In mice, the first weeks of the postnatal period are crucial for the development and the maturation of muscle. This critical window is characterized by significant muscle growth, which occurs through hypertrophy of existing muscle fibers rather than the formation of new ones (hyperplasia). Myonuclear accretion by satellite cells fusing into the growing myofibers plays an essential role in this process. In the mouse extensor digitorum longus (EDL), myofiber cross-sectional area increases threefold between postnatal day 7 (P7) and day 21 (P21), accompanied by a 2.4-fold increase in the number of myonuclei per fiber and a corresponding threefold decrease in the number of satellite cells per fiber. 24 Between P21 and P56, the myofiber size continues to increase up to ∼2.5-fold, while the increase in number of myonuclei per fiber is more limited, resulting in an expansion of the myonuclear domain.24,25 Analysis of satellite cell activity showed that a high proportion of satellite cells (∼65%) actively divide at birth and that this rate decreases by half by P21, reaching near-complete quiescence by P56. 26 The expression of sex hormones in male and female mice (testosterone and estradiol, respectively) at the pubertal stage was shown to activate Notch signaling and promote cell cycle exit, thereby contributing to satellite cell quiescence. 27
Satellite cell depletion using Pax7cre-diphtheria toxin (DTA) mice showed that prepubertal depletion impairs myofiber growth and myonuclear accretion, underlying the importance of satellite cells in early post-natal development. 25 In the absence of injury, the fusion of satellite cells into myofiber is strongly reduced after 8-12 weeks of age, although spontaneous fusion is still observable in resting muscles in adult mice.25,28 Depletion of satellite cells in uninjured adult or aged mice does not induce myofiber atrophy per se, but is associated with an increase in collagen content. 29 However, ablation of satellite cells blunted muscle adaptation in response to overload hypertrophy and exercise training.30,31 Analysis of satellite cell depletion in a model synergist ablation overload in young adult mice (2 months old) vs mature adult mice (4 months old) showed that muscle hypertrophy was prevented only in young adult mice, suggesting that the reliance on satellite cells depends on the maturation age. 32 In addition to myonuclear accretion, it was shown that satellite cells communicate with mononucleated cells and myofibers through extracellular vesicles to coordinate the hypertrophic response and extracellular matrix (ECM) production, which is critical for effective muscle growth. 33
Adult myogenesis
Adult skeletal muscle possesses a remarkable regenerative capacity, restoring myofiber integrity and function after severe injury within a few weeks in mice, although this timescale may be extended in humans.34,35 Ablation studies have shown that satellite cells are absolutely essential for this regenerative process. 36 Notably, the efficiency of muscle regeneration is strongly influenced by the integrity of the ECM; its disruption, as observed in volumetric muscle loss, results in a failure of muscle regeneration and replacement of the lost tissue with fibrotic scar.37,38
In resting adult skeletal muscle, satellite cells are mitotically quiescent but extend long cytoplasmic projections, which serve to monitor biomechanical or signaling changes in the myofiber or its microenvironment.39,40 Satellite cells respond to molecules called DAMPs (Damage-associated molecular patterns) released by damaged fibers or neighboring cells (e.g., fibroadipogenic progenitors or resident macrophages) into the extracellular environment. 41 It was shown that distant injury (contralateral muscle) induced systemic signalling, such as HGF (Hepatocyte Growth Factor), that primes satellite cells to enter a GAlert stage characterized by elevated mitochondrial activity, faster cell cycle entry, and phosphorylation of ribosomal protein S6 (RPS6). 42 In the injured mouse muscle, satellite cells enter the cell cycle and perform the first cell division within 40-48 h, and subsequent cell divisions in 〜10 h. 42 At 〜4 days post-injury myoblasts start to the exit cell cycle to differentiate and fuse into regenerating myofibers.43,44 Satellite cells reacquire quiescence mainly between 5-10 days post-injury, although a subset of cells also return to quiescence earlier. 45 In humans, the kinetics of satellite cell activation and proliferation are slower than in mice. When isolated and cultured in vitro, human primary myoblasts typically undergo their first division after 50–60 h and subsequent divisions in 〜20 h, 46 whereas satellite cells within intact human muscle fibers divide after approximately 3–4 days in culture. 47
Different paired box proteins, MRFs, and signalling pathways are essential for cell fate decisions during adult myogenesis. Conditional deletion of Pax7 by tamoxifen injection in adult Pax7flox/CreERT2 mice revealed the lack of this transcription factor induced cell cycle arrest in satellite cells and blocked their ability to form regenerating fibers, resulting in fibrofatty replacement. 48 Notch signaling is a key regulator of Pax7, 49 and conditional deletion of Notch1 and/or Notch2 in satellite cells strongly impairs their expansion and self-renewal, resulting in impaired myogenesis. 50 MRFs like MRF4, Myf5 and MyoD also play a key role in this process. Knockout of Mrf4 was shown to reduce myokine production and lead to spontaneous activation of satellite cells and progressive reduction of the satellite cell pool. 51 Conditional knockout of both Myf5 and MyoD did not affect the satellite cell pool or the muscle phenotype in the absence of injury; however, the regenerative response is strongly impaired leading to fibrosis and fat accumulation. 52 Mice knockout for Myf5 and carrying only one functional MyoD allele did not show striking regenerative defects, while mice knockout for MyoD and carrying a single functional Myf5 allele were able to regenerate although this process was impaired. 52 These results suggest that there are compensation mechanisms between Myf5 and MyoD. Noteworthy, a subset of satellite cells (∼10%) never expressed Myf5 during development and these cells display deeper quiescence and higher self-renewing capacity through asymmetric division than Pax7+/Myf5 + cells that are more prone to myogenic commitment.53,54 This heterogeneity in the adult satellite cell pool revealing different myogenesis capacity and dynamics has been observed using different models.55–57
Conditional knockout of Myomaker or Myomixer in satellite cells did not affect the early regenerative response (at 3 days post-injury), but strongly impaired myofiber formation at later time points indicating impaired fusogenic capacity.58,59 At these later myogenic stages, there is a switch from Notch signalling to the activation of the Wnt pathway that is needed to promote myoblasts fusion and myogenic lineage progression. 60 Conversely, the TGF-β signalling pathway is a negative regulator of myogenic cell fusion that prevents active cytoskeletal remodeling.61,62 The myogenesis process is completed with the maturation of myofibers and the expression of contractile proteins such as MyHC. Loss of Myh3 or Myh8 was shown to affect satellite cell pool, muscle fiber size and/or phenotype, and increase fibrosis.63,64
Altogether, these findings indicate that satellite cells are critical for muscle development, post-natal growth and adult muscle regeneration. Loss of function of key transcription factors and signalling pathways could affect satellite cell function and impair myogenesis leading to myopathies.
Pathophysiology of cell-opathies
Recent work from the Zammit lab has laid the groundwork for the emerging field of satellite cell dysfunction in various myopathies, a concept they introduced as satellite cell-opathies.2,3 This term describes a pathological condition in which the dysfunction, depletion, or misregulation of satellite cells contributes significantly to the progression of muscle disease. These myopathies are classified into two types: primary satellite cell-opathies that primarily impair satellite cell function, and secondary satellite cell-opathies that affect both satellite cells and myofibers. By distinguishing whether satellite cells lie at the core of the pathophysiological mechanism or contribute to the pathogenesis through parallel processes, this concept provides a framework to better understand muscle regeneration deficits in myopathies (Figure 1). In the following section, we will explore these myopathies in more detail.
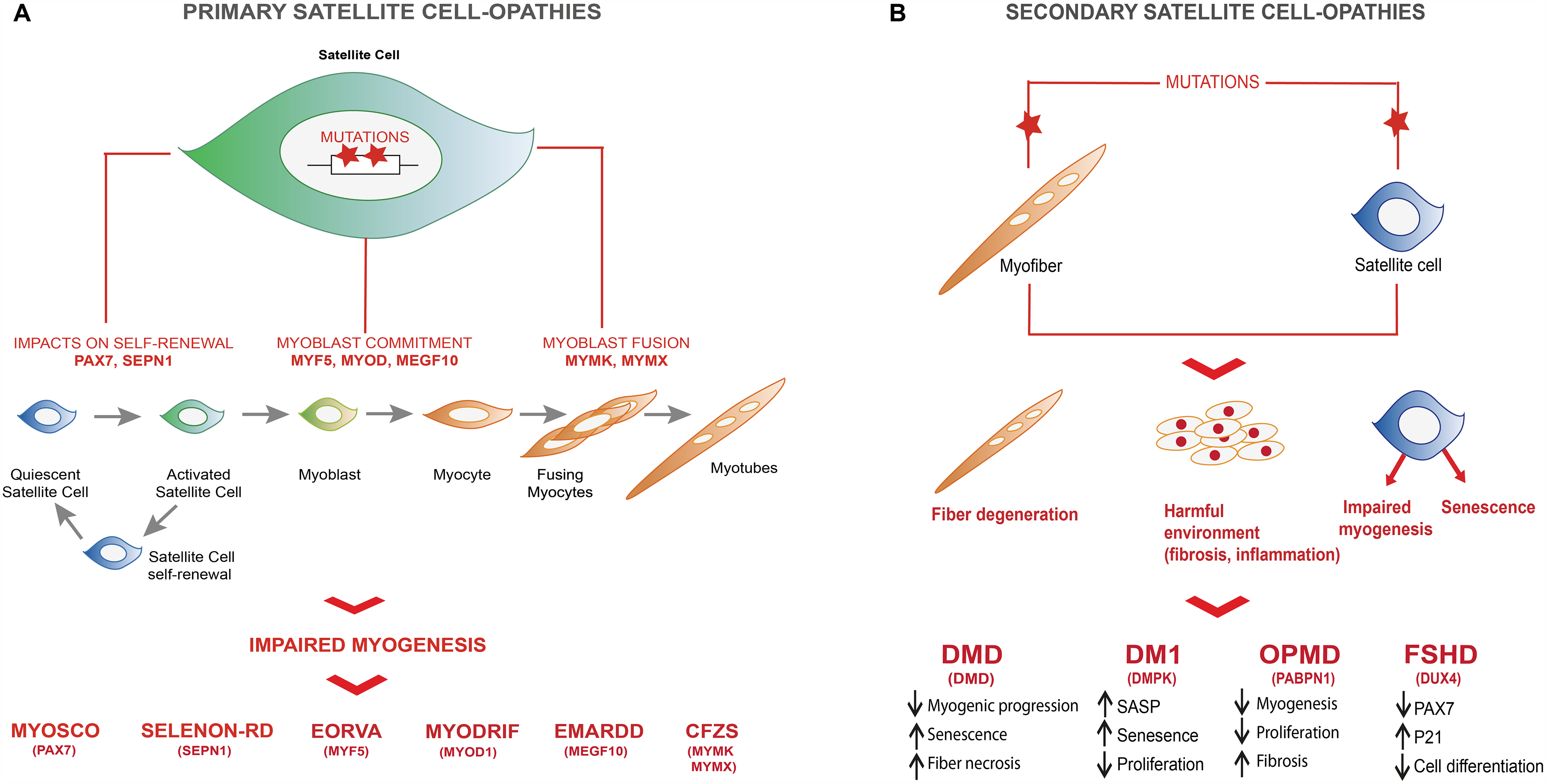
Primary and secondary satellite cell-opathies. (
Primary satellite cell-opathies
Altogether, these findings demonstrate that an increasing number of diseases primarily affect satellite cells, leading to severe myopathies without directly impacting the myofibers per se.
Secondary cell-opathies
While this is not an exhaustive list of all the satellite cell-opathies, it highlights the previously underappreciated role of satellite cell dysfunction in myopathies. Further studies are needed to investigate satellite cell defects in other muscular diseases and to determine their contribution to disease progression.
Regenerative therapies for satellite cell-opathies
Current therapies for myopathies primarily focus on repairing dysfunctional myofibers or mitigating their detrimental microenvironment, such as chronic inflammation and fibrosis. However, with the growing recognition of satellite cell-opathies, there is a pressing need to develop therapeutic strategies that specifically target defective satellite cells. Cell-based therapies, such as satellite cell transplantation, induced-pluripotent stem cells (iPSCs), organoids, and bioengineering tissues, are attractive strategies aiming to restore muscle function by replenishing the pool of functional satellite cells (Figure 2). Other key strategies in this area include targeted gene therapy, drug-based precision medicine, and exercise (Figure 2). This next section aims to explore the therapeutic potential of these strategies for the treatment of satellite cell-opathies. All therapies discussed in this section are summarized in Table 1.
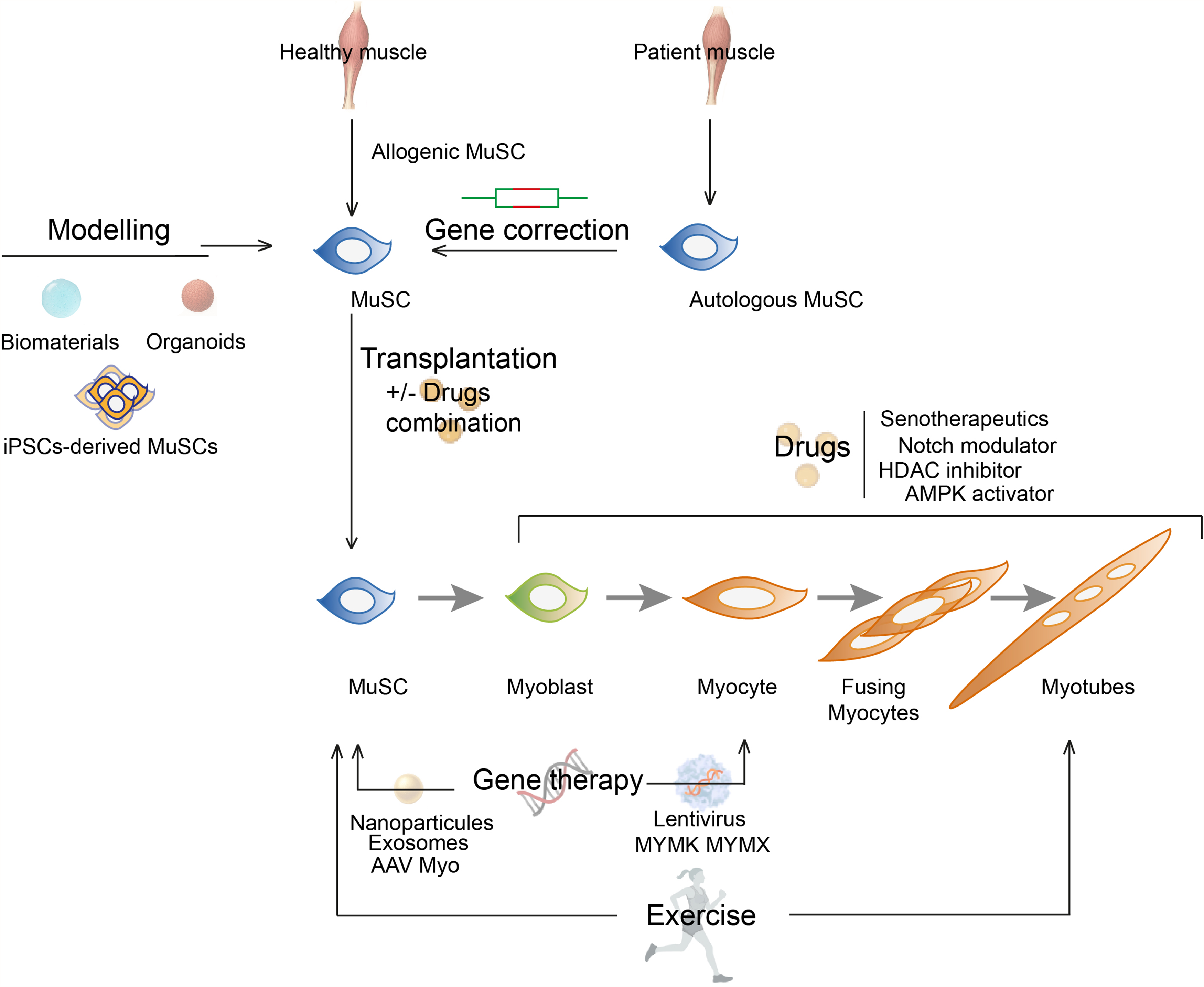
Regenerative therapies for satellite cell-opathies. Schematic showing different potential therapeutic approaches targeting satellite cells for the treatment of satellite cell-opathies: cell-based therapies, drug-based therapies, gene therapies, and exercise. The combination of multiple approaches is a promising avenue to target the complex pathogenic mechanisms associated with satellite cell-opathies.
Summary of therapeutic strategies for satellite cell-opathies and their advantages and limitations.

Cell transplantation
Cell transplantation is a promising therapeutic approach for satellite cell-opathies because it addresses the fundamental issue in these disorders: the dysfunction or depletion of satellite cells, which are essential for muscle regeneration. Allogenic satellite cell transplantation involves isolating satellite cells from healthy donor muscle tissue and transplanting them into the patient's muscle to enhance regeneration, while autologous transplantation usually requires genetic correction of the patient's cells prior to transplantation, depending on the disease context. This approach has been shown to be feasible in both human and mouse models, although it demonstrated more effective results in mice.163–165 Transplantation of human myoblasts into immunodeficient mice has been shown to result in low proliferation and migration, along with premature differentiation. 166 Upon transplantation, mouse satellite cells contribute to muscle repair by fusing with existing fibers and can also engraft in the niche to self-renew and sustain long-term regeneration.167,168 A subset of satellite cells with higher stemness capacity was shown to have higher long-term engraftment and self-renewal capacity.54,57 In particular, transplantation has shown promise in the context of muscular dystrophy, such as DMD, to restore dystrophin expression in myofibers.127,131,169 More recently, studies have focused on diaphragm transplantation in DMD models, highlighting its therapeutic potential for improving muscle function. 128 Considering the critical role of dystrophin in satellite cell function, transplanted healthy cells could also help mitigate the myogenic defects observed in DMD.
A critical factor for successful satellite cell engraftment is the preservation of an intact niche unoccupied by host satellite cells. It was shown that the efficacy of satellite cell transplantation is strongly enhanced after irradiation-induced deletion of host satellite cells. 170 This has important implications, particularly for primary satellite cell-opathies such as MYOSCO, where the exhaustion of the satellite cell pool may significantly enhance the success of transplantation compared to what has been observed in other myopathies. Additionally, different approaches have demonstrated that a short-term exposure of freshly isolated satellite cells to molecules like prostaglandin-E2, Tubastatin A or the RTK inhibitor CEP-701 enhances their long-term regenerative capacity upon transplantation.171,183,184
Secondary satellite cell-opathies that affect predominantly a specific group of muscles, such as OPMD, may also represent promising targets for cell-based therapies. This approach has already been tested in a Phase I/IIa clinical trial involving 12 genetically confirmed OPMD patients who underwent autologous myoblast transplantation following cricopharyngeal myotomy. 130 The study confirmed the feasibility, tolerance, and safety of the therapy after more than 2 years of follow up. Further studies are needed to evaluate the clinical efficacy of this approach in OPMD and in the broader context of satellite cell-opathies. 185
Despite the promising advances in satellite cell-based therapies, there are significant challenges that remain. The repeated cycle of degeneration and regeneration observed in certain muscular dystrophies can exhaust the satellite cell pool and severely limit their proliferative capacity in vitro, rendering them poorly suited for genetic correction and for autologous transplantation to generate sufficient genetically corrected myofibers to meaningfully improve muscle function. Conversely, transplantation of allogenic satellite cells is challenged by immune rejection, which not only prevents long-term engraftment but can also exacerbate muscle pathology through bystander effects, including inflammation and damage to neighboring fibers. 172 Limited cell availability, poor cell migration, lack of a systemic administration method, and immune rejection are among the key hurdles that hinder the widespread clinical application of these therapies. This explains why clinical trials using satellite cell or myoblast transplantation for the treatment of myopathies have resulted in limited clinical improvements. 186 These considerations highlight the need for alternative strategies, such as improved cell expansion methods, priming of satellite cells ex vivo with growth factors, or combination approaches integrating pharmacological modulation of the satellite cell niche to enhance engraftment and regenerative potential. Despite the challenges and the limited therapeutic benefits observed to date, primary satellite cell-opathies with a compromised satellite cell pool, as well as secondary satellite cell-opathies affecting a limited number of muscles (e.g., OPMD), appear to be ideal candidates for definitively testing the efficacy cell transplantation under optimal conditions, an avenue that remains poorly explored.
Induced pluripotent stem cells (iPSCs)
Technological advancements, particularly the reprogramming of somatic cells (e.g., peripheral blood mononuclear cells, skin fibroblasts) into iPSCs, 187 can overcome many of the limitations associated with satellite cell transplantation and offer an innovative approach for treating both primary and secondary satellite cell-opathies. iPSCs can be differentiated into myogenic progenitors either through transient PAX7 or MYOD1 overexpression, or by presomitic mesoderm induction using combinations of small molecules modulating developmental pathways, such as the GSK-3 inhibitor CHIR99021.188–190 These myogenic progenitors have the potential to replenish the satellite cell niche, improving muscle repair in conditions where satellite cell function is compromised, such as LGMDs.191–193
The advent of human iPSCs has provided a powerful platform for developing regenerative therapies.132,194,195 Recent advances have enabled the efficient, transgene-free differentiation of iPSCs into striated muscle fibers and satellite-like progenitors through modulation of Wnt and BMP signaling pathways, supporting applications in disease modeling, drug screening, and therapeutic development. 133 The identification of surface markers such as ERBB3 (Erb-B2 Receptor Tyrosine Kinase 3) and NGFR (Nerve Growth Factor Receptor) has further refined the isolation of functional PAX7+ myogenic progenitors, facilitating gene correction strategies, such as clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9–mediated dystrophin restoration in DMD models, and in vivo engraftment. 173 Recently, the feasibility of generating satellite cells from CRISPR-Cas9-corrected iPSCs has been demonstrated in both allogeneic and xenogeneic hosts. 196 These chimeric models produced functional satellite cells capable of restoring dystrophin expression in DMD mice, offering a promising strategy for generating human satellite cells from large animal models that could be leveraged for regenerative medicine applications in humans. The use of iPSCs offers several advantages, including the potential for an almost unlimited cell source. It was shown that iPSCs could be expanded as much as 5 × 1011-fold and still maintain their ability to form myotubes. 195 This is particularly important in the context of myogenic cell transplantation, where a large number of cells are required to target widespread muscle tissue, and where freshly isolated satellite cells rapidly lose their stemness and engraftment potential when expanded in vitro. 168 Moreover, iPSCs provide a platform for genetic correction prior to differentiation, enabling the autologous correction of the patient's cells with genetic muscle disorders such as DMD. When derived from the patient, iPSCs also maintain immunological compatibility, reducing the risk of rejection. Overall, iPSC-based approaches offer greater scalability, flexibility, and potential for disease correction compared to direct transplantation of primary somatic cells.188,194 Recently, the first off-the-shelf, clinical-grade iPSC-based therapy was developed to regenerate both muscle tissue and its stem cell niche, with validated safety and efficacy in animals, and FDA (U.S. Food and Drug Administration) authorization for clinical use in DMD. 197
However, several challenges must be overcome before iPSC-based therapies can be widely applied in clinical settings. One major concern is tumorigenicity, as incomplete differentiation of iPSCs may lead to tumor formation. Additionally, achieving highly efficient and reproducible myogenic differentiation remains a significant technical challenge, which could impact the therapeutic applicability of these cells. Addressing these limitations is crucial to unlocking the full potential of iPSCs in regenerative therapies for satellite cell-opathies. 198 One approach currently investigated is the introduction of failsafe systems, such as incorporation suicide gene, that could be triggered to remove or inactivate the transplanted cells if needed. 199 Another avenue is the development of fully characterized iPSC lines that can serve as universal donor cell sources for transplantation, with demonstrated safety, scalability, reproducibility, and immune tolerance by the host. 200 However, a major translational challenge is that differentiated cells derived from hiPSCs are not inherently autologous and can be recognized as foreign by the patient's immune system, leading to immune rejection. One proposed strategy to overcome this barrier is the development of Human leukocyte antigen (HLA)-matched hiPSC banks, which would allow selection of lines compatible with a patient's immune profile.201–203 While theoretically feasible, establishing and maintaining such a bank would require extensive resources, rigorous quality control, and coverage of population HLA diversity, rendering it very expensive. These economic considerations are critical, as the high cost and logistical complexity of some hiPSC-based approaches could limit their clinical translation, despite their scientific potential. Detailed pharmacoeconomic analyses are needed to ensure that the cost–benefit balance for patient care is optimized, supporting not only biological efficacy but also practical feasibility and long-term sustainability.
Overall, iPSCs represent a virtually unlimited source of patient-specific or universal donor cells that may help address key challenges in cell transplantation for satellite cell-opathies. However, the success of such approaches also depends on the presence of a supportive environment, an aspect that will be further discussed in the next section.
Biomaterial-based approaches
Biomaterials are critical in creating environments that mimic natural muscle tissue, providing the necessary support for satellite cells or iPSC-derived myoblasts. Many components of the ECM, such as fibronectin, laminin, and different types of collagens, were shown to play a role in vivo in the regulation of satellite cell function.204–206 Scaffolds, hydrogels, and ECM mimics are utilized to recreate these cues, facilitating more efficient satellite cell engraftment and muscle regeneration.207,208 Although biomaterial-based strategies have been primarily developed in the context of volumetric muscle loss (VML), many of these approaches are equally relevant to satellite cell-opathies, where restoring or protecting the satellite cell niche is essential for improving regenerative capacity. Many different types of hydrogels have been tested in the field (see review in Lev and Seliktar 209 ). For instance, one approach involves the use of hyaluronan and methylcellulose hydrogel as a delivery system to enhance satellite cell transplantation by increasing donor cell proliferation, reducing clearance, and promoting fiber dispersion, ultimately improving muscle repair. 210
A key challenge in expanding and transplanting satellite cells is creating an environment that supports their migration, proliferation, and differentiation. Biomaterial scaffolds, such as hydrogels and decellularized ECM, offer a 3D structure that enables satellite cells to maintain their functionality while integrating into damaged muscle tissue.211–213 These scaffolds can also be loaded with bioactive molecules like FGF-2 (Fibroblast Growth Factor 2) and IGF-1 (Insulin-like Growth Factor 1), which directly stimulate satellite cell activation, thereby promoting muscle repair. 214 In addition to their biochemical composition, the physical properties of biomaterials, particularly stiffness, play a critical role in regulating satellite cell fate and myogenicity. Myogenic cells cultured on a soft hydrogel that mimics the elasticity of the muscle showed better self-renewal and engraftment upon transplantation. 215
In satellite cell-opathies like DMD, hydrogels not only support muscle tissue regeneration but also protect satellite cells from oxidative stress and inflammation, which can impair their function. For instance, a hydrogel-based system was developed and tested in DMD models, where it enhanced satellite cell survival and engraftment, even in non-irradiated and immunocompetent mice. 216
Delivery of these biomaterial scaffolds is a potential limitation for therapeutic applicability. To this end, an injectable biomimetic strategy was developed by engineering a peptide amphiphile-based liquid crystal scaffold that aligns with host myofibers, retains growth factors, and promotes myogenic progenitor cell engraftment. 217
Biomaterial-based approaches cannot address all the technical issues associated with cell transplantation, such as immune rejection; however, technological advancements, particularly 3D bioprinting, offer promising bioengineered solutions for some of these challenges by enabling the creation of customized scaffolds with complex architecture that facilitates satellite cell expansion and/or engraftment. 218 Although this strategy is largely investigated for VML, the use of muscle tissue constructs preloaded with satellite cells in their native niche may enhance cell engraftment and hold therapeutic potential in satellite cell-opathies. 219
Organoids
A critical component of the satellite cell niche is their interaction with other cell types, such as vascular cells, immune cells, and fibroadipogenic progenitors. In this context, organoids represent a promising tool to co-culture myogenic progenitor cells with other cell types in a 3D environment, which offers a potential solution to improve the efficacy of cell transplantation in satellite cell-opathies.174,220 In current therapeutic strategies, these organoid systems are used primarily as platforms to expand satellite cells in vitro while preserving stemness and engraftment potential, rather than as transplantable structures themselves.
A recent breakthrough in this field involved generating in vitro-derived satellite cells (idSCs) from skeletal muscle tissue, addressing the limitations of earlier cell therapies that used committed myogenic progenitors rather than true satellite cells. Myogenic cells expanded in a spinner flask gave rise to 3D skeletal muscle organoids, which differentiated into myotubes and simultaneously generated a self-renewing satellite cell-like population. 221 When transplanted into injured mice, idSCs successfully integrated into muscle fibers, repopulated the satellite cell niche, self-renewed, and enhanced muscle regeneration and force production, demonstrating functionality superior to myoblasts and comparable to native satellite cells. Moreover, when dystrophic mdx mice were depleted of their host satellite cells by irradiation, the long-term engraftment of this satellite cell-like population and formation of dystrophin-positive myofibers is enhanced compared to freshly isolated satellite cells. 221 This suggests that such a therapy may be particularly well suited for satellite cell-opathies in which satellite cells are dysfunctional and/or depleted. Notably, this work has been validated in both human and mouse models, further underscoring the potential of idSCs as a scalable therapeutic option for genetic muscle disorders like satellite cell-opathies.
Another model testing different cell types to form heterotypic 3D embryoids showed that the combination of embryonic endothelial cells and fibroblasts strongly supported the myogenic specification of human iPSCs. 134 iPSC-derived myogenic cells displayed higher Pax7 expression and self-renewal capacity. Moreover, transplantation of these myogenic cells in dystrophic mice showed higher engraftment capacity and enhanced muscle function compared to primary myoblasts.
Altogether, these findings highlight the critical role of the cellular microenvironment to optimize satellite cell expansion and engraftment for cell therapy. Although 3D organoids hold promise for cell-based therapies, they do not fully resolve the challenges inherent to autologous cell use in satellite cell-opathies. Their full potential can be unlocked by gene therapy, providing precise genetic corrections that restore cell functionality and durable therapeutic benefit.
Gene therapy
Gene therapy holds great potential for satellite cell-opathies by providing durable muscle repair via correction of the underlying genetic defect. Because satellite cells are self-renewing, genetic modifications can provide lasting benefits in muscle regeneration. This approach is particularly effective for monogenic disorders like DMD, where advances in adeno-associated virus (AAVs) have successfully restored dystrophin expression in myofibers of Mdx4cv mice135–137 and canine models. 138 Clinical trials using a single intravenous injection of AAV carrying microdystrophin have demonstrated its ability to induce microdystrophin expression in skeletal muscle. 142 While the primary endpoint (North Star Ambulatory Assessment) was not met, other secondary functional endpoints (e.g., time to rise, 10-meter walk) showed positive trends. Importantly, this clinical trial used the rAAVrh74 vector, which showed lower seroprevalence compared to other AAVs (e.g., AAV2, AAV8), minimizing the risk of immune reaction. 222
Beyond DMD, gene editing has been applied to other muscular dystrophies, including DM1, where CRISPR interference has been used to target toxic CUG repeats in the DMPK gene. 175 Reduction of CUG-expanded DMPK transcripts partially corrected splicing abnormalities and foci formation, thereby restoring physiological parameters in DM1 myotubes. 139 Similarly, FSHD gene therapies have focused on silencing the DUX4 gene 140 and employing miRNA-based strategies. 223 Recent studies have also leveraged CRISPR-Cas9 technology in iPSCs, allowing genetically corrected cells to be engrafted and restore muscle function in disease models, such as DMD.224,225
By directly targeting the underlying genetic defect, CRISPR-based approaches offer the potential for durable, curative interventions; 226 however, major limitations currently constrain their therapeutic application. First, gene delivery and editing efficiency remains suboptimal, particularly in quiescent satellite cells and post-mitotic myofibers, meaning that only a fraction of target cells is successfully corrected. Second, off-target effects create a risk of undesirable mutations and long-term safety concerns. Ongoing technological developments, including high-fidelity Cas variants, improved guide RNA design, and optimized delivery systems, are expected to enhance both specificity and efficiency.
Gene therapies have primarily focused on correcting gene expression in myofibers; however, treating satellite cell-opathies will require the specific targeting of satellite cells. These cells are particularly challenging to target due to their small size, rarity, and quiescent state. A study screening different types of AAVs has shown that systemic injection of AAV9 was more efficient than others (e.g., AAV1, AAV6.2) to target satellite cells. 176 This efficacy was greater in mdx mice, where satellite cells are activated due to ongoing muscle degeneration, compared to wildtype mice, in which satellite cells remain predominantly quiescent. A sustained dystrophin expression was observed following repeated injury, highlighting the long-term gene contribution of dystrophin-corrected satellite cells. 176 Recent advances in vector engineering, including the development of muscle-tropic capsids such as AAVMYO, AAVMYO2, and MyoAAVs, have enhanced transduction efficiency in satellite cells from mature muscle tissue, but this efficacy remains partial, especially for systemic delivery, highlighting the challenge of targeting satellite cells.141,227 It was shown that AAV8, AAVMYO, and AAVMYO2 were able to deliver transgenes to different mononuclear cells in muscle including MuSCs, both in healthy and LAMA2-related muscular dystrophy (dyW/dyW) mice, providing valuable insights for designing gene therapies targeting MuSCs in context of dystrophies. 141 Another groundbreaking approach showed that engineering of lentiviruses pseudotyped with Myomaker and Myomerger (Myomixer) could be used to transduce specifically myofibers and myogenic progenitors. 228
While gene therapy holds great promise for treating genetic disorders, its success largely depends on safe and efficient gene delivery. Systemic administration of AAVs is largely uptaken by the liver, decreasing the vector bioavailability and contributing to hepatotoxicity. 229 Immune response is also an important safety concern. A recent N-of-1 study using a recombinant AAV9 vector encoding a transcriptional activator (VP64 fused to dead Staphylococcus aureus Cas9) to upregulate a non-muscle full-length isoform of dystrophin triggered an innate immune reaction resulting in acute respiratory distress syndrome and cardiac arrest in an adult DMD patient. 230 In addition to the immune response to the vector used, it has also been shown that the immune system can react to the dystrophin protein itself leading to the formation of autoreactive dystrophin-specific T cells. 231
Moreover, the efficacy of AAV delivery is significantly reduced in dystrophic muscle due to ongoing cycles of myofiber degeneration and regeneration, which lead to the loss of viral particles before they can achieve stable transduction, as well as reduced transgene transcription due to promoter silencing or modified transcriptional activity of the regenerating myonuclei. 177 Consequently, these findings underscore a strong rationale for combined therapeutic strategies, where AAV-mediated gene delivery could be complemented with approaches that stabilize myofibers, dampen the chronic inflammation and fibrosis, modulate satellite cell function, or enhance muscle regeneration, thereby increasing the window of opportunity for effective transduction. Integrating gene therapy with pharmacological agents, cell-based therapies, or interventions targeting the muscle niche may therefore optimize long-term therapeutic benefit in dystrophic conditions.
Altogether, these findings underscore the urgent need to develop safer, more efficient, and satellite cell-specific delivery methods, which will be critical for advancing therapies targeting both primary and secondary satellite cell-opathies.
Nanoparticles
Nanoparticles have emerged as a promising tool for targeted therapeutic delivery in satellite cells and dystrophic muscles. Gold nanoparticles have been explored as carriers for small oligonucleotide sequences to compensate for dystrophin deficiency in DMD. 143 By conjugating an aptamer targeting α7B1 integrin, these nanoparticles enable the direct delivery of microRNA-206 into satellite cells, leading to improved muscle functionality in a DMD mouse model. In addition, nanoparticles containing a PTEN (phosphatase and tensin homolog) inhibitor have been shown to restore muscle function in DMD by promoting muscle regeneration and reducing fibrosis. 144
Beyond gold-based systems, lipid nanoparticles have demonstrated the ability to efficiently deliver Cas9 mRNA and sgRNA into skeletal muscle, advancing gene-editing approaches for muscular dystrophy. 145 More recently, exosome-based therapies have gained attention as an alternative delivery method, offering a biocompatible and efficient platform for therapeutic cargo transport.178,232 Myo-exosomes were engineered as ferromagnetic nanocarriers that could be directed toward skeletal muscle after systemic injection using an external magnetic field. However, they were found to preferentially target macrophages, thereby reducing the inflammatory response and improving muscle function in mdx mice, rather than specifically targeting satellite cells. 232
Additionally, the development of NanoCas technology represents a new frontier in gene therapy, with the potential to bypass hepatic sequestration of AAVs. This approach has shown markedly enhanced targeting of skeletal muscle in a DMD model. 129 Insights gained from this disease are expected to pave the way for the development of safe and effective treatments targeting satellite cells, an urgent need for addressing satellite cell-opathies.
Drug based therapy
As discussed previously, the delivery and biodistribution of gene therapy vectors remain major challenges. Satellite cells reside beneath the basal lamina and are in a quiescent state in healthy muscle, which makes them difficult for viral vectors to access due to the anatomical protection of the satellite cell niche or transduce effectively. In contrast, pharmacological approaches can rapidly penetrate muscle tissue, act on both quiescent and activated cells, and modulate key signaling pathways governing proliferation and differentiation without the need for efficient intracellular delivery or receptor-mediated uptake. Moreover, small-molecule therapies can be repeatedly administered and reversed, whereas viral delivery is difficult to repeat and produces permanent effects, including potential side effects.
Small molecules can target intrinsic defects in satellite cells and/or extrinsic factors that create an unfavorable muscle environment for satellite cells (e.g., excessive fibrosis, chronic inflammation). There is emerging evidence that, in addition to genetic satellite cell-opathies, this condition can also be non-hereditary. Acquired satellite cell-opathies result from environmental or systemic factors associated with aging, chronic inflammation, metabolic disorders, or prolonged muscle disuse, which in turn alter the satellite cell niche and impair their regenerative potential. Acquired satellite cell-opathies may be particularly amenable for pharmacological modulation of the muscle environment. 233 Among the promising pharmacological strategies explored for inherited or acquired satellite cell-opathies, Notch signaling modulators, metabolic modulators and senotherapeutics hold strong potential.
Notch signaling plays a crucial role in satellite cell activation and self-renewal,49,234 and its dysregulation contributes to some primary satellite cell myopathies. Activating Notch signaling in myofibers upregulates their expression of the Notch ligands Delta-like 1 and Jag1 which promoted satellite cell self-renewal. 179 Accordingly, overactivating Notch signalling was shown to ameliorate the muscle phenotype in different dystrophic models.146,179 Drugs targeting the Notch regulator AAK1 (Adaptor-associated protein kinase 1) showed the potential to restore cell polarity and have promising therapeutic benefits in DMD models. 235 Other drugs targeting the Notch pathway, such as valproic acid, showed a capacity to enhance myogenesis.180,236 Valproic acid is also known as a histone deacetylase (HDAC) inhibitor, a class of drugs that modulate gene expression by enhancing chromatin accessibility, and are extensively studied for the treatment of myopathies. The HDAC inhibitor Givinostat showed promising capacity to reduce symptoms progression in a clinical trial with DMD patients. 157 HDAC6 modulators have been found to counteract myofiber dysfunction by modulating TGF-β signaling and reducing fibrosis in dystrophic mouse models.147,148 Since TGF-β is also known to repress myogenesis and muscle regeneration, 61 modulating its activity represents an attractive target for satellite cell-opathies. Accordingly, HDAC inhibitors, such as vorinostat also showed therapeutic potential for other satellite cell-opathies, like DM1. 150
Another therapeutic approach showed that targeting the satellite cell niche, especially the vasculature, holds therapeutic potential for multiple muscular dystrophies in which satellite cell function is impaired. 149 Apelin peptide treatment restored satellite cell function through the release of angiocrine factors in models of DMD, Laminin-α2, and Collagen-VI myopathies, 149 highlighting the importance of the surrounding microenvironment in satellite cell function.
In addition to signaling modulation, restoring energy balance in satellite cells has emerged as a key strategy for treating muscle-wasting conditions. AICAR (5-Aminoimidazole-4-carboxamide ribonucleotide), an activator of AMP-activated protein kinase (AMPK), has shown potential due to its role in enhancing mitochondrial biogenesis, glucose uptake, and endurance. 152 AMPK activation also reduces inflammation and fibrosis, contributing to better muscle integrity. This metabolic approach is particularly relevant in DM1, where studies have highlighted impaired AMPK activation and persistent mTORC1 activity as major contributors to muscle dysfunction. 153 Pharmacological activation of AMPK with AICAR has demonstrated multiple benefits, including reducing myotonia, improving muscle histology, and partially correcting RNA mis-splicing.154,237 Additionally, mTORC1 inhibition with rapamycin has been shown to enhance muscle relaxation and force without affecting RNA splicing. Further supporting the role of metabolic regulation, recent studies have revealed systemic impairments in oxidative phosphorylation, ATP production, and mitochondrial function in DM1, alongside increased ROS accumulation. Notably, metformin treatment has been found to reverse mitochondrial dysfunction, restore energy metabolism, and improve cellular proliferation defects in DM1 cells113,151 reinforcing the potential of metabolic interventions in satellite cell-related disorders.
As described earlier, another hallmark of satellite cell-opathies is cellular senescence. 238 Senescent cells express high levels of SASP contributing to disease progression.109,181 In this context, senotherapeutic drugs that target senescent cells and/or their production of SASP have gained interest as potential treatments 239 . For example, the senolytic combination of dasatinib and quercetin has been shown to improve muscle function in aged mice by selectively eliminating senescent cells.178,181 Different senotherapeutic drugs were successfully tested in preclinical models of DM1 155 and DMD, 156 however, these findings have not yet been confirmed in human patients, which will be critical given the different mechanisms of cellular senescence and the different lifespans in human compared to mice. 240 The first clinical trials using senolytics for conditions such as Alzheimer's disease or idiopathic pulmonary fibrosis suggest that these drugs are safe and tolerable, but the functional impact remains to be determined. 241 More recently, researchers have identified JUNB as a key transcriptional regulator of SASP factors in senescent satellite cells, suggesting that targeting this factor could modulate the senescence program and enhance muscle regeneration. 242 Inhibition of the IKK/NF-κB pathway using SR12343 has shown promise in reducing signs of senescence and muscle degeneration, and improving muscle strength. 243
Together, these pharmacological approaches highlight the potential of targeting satellite cell dysfunction to treat satellite cell-opathies. Continued research and clinical trials will be essential to translate these findings into effective treatments for patients with satellite cell-related muscle disorders.
Exercise
Physical activity plays a well-documented role in maintaining muscle health and function. In myopathies, exercise is often considered a double-edged sword, as it may exacerbate muscle damage due to increased oxidative stress and impaired muscle repair. However, recent studies challenge this notion, with growing evidence showing that adapted exercise regimens promote satellite cell activation, improve muscle strength, and enhance regenerative capacity in satellite cell-opathies like DM1 and LGMD.161,162,182,244 Moreover, in patients with FSHD, a recent study showed that 24 weeks of combined aerobic and resistance training increased the number of satellite cells, particularly in fast fibers, without worsening muscle histopathology or inflammation. Importantly, telomere length was preserved, indicating that training enhanced regenerative potential without promoting replicative exhaustion. 160 Even in muscular dystrophies characterized by myofiber fragility and recurrent degeneration, such as DMD, emerging evidence suggests that exercise can improve muscle strength and endurance in patients, challenging the long-standing assumption that physical activity is necessarily detrimental in these conditions. 159 Nonetheless, exercise training must be adapted and carefully monitored to minimize the risk of additional damage.
One key benefit of exercise is its ability to stimulate mitochondrial metabolism, which is impaired in many muscular diseases like DM1. 158 Studies using treadmill exercise in DM1 mouse model have demonstrated its role in promoting mitochondrial fusion and fission, processes essential for maintaining skeletal muscle function. Additionally, research exploring the therapeutic potential of AMPK activation in DM1 has shown that combining AICAR treatment with swimming exercise leads to a greater reduction in toxic CUG RNA foci, partial reversal of MBNL1 sequestration, and correction of pathogenic splicing defects. 152 This combined intervention also promotes muscle fiber hypertrophy, further highlighting the therapeutic value of exercise in DM1. While exercise has been shown to be effective for DM1 pathology, further investigations on other satellite cell-opathies, notably the primary ones, are needed to determine their potential benefits.159,245
Conclusion and perspectives
In this review, we have discussed the underlying mechanisms of satellite cell impairments in both primary and secondary satellite cell-opathies and their contribution to disease progression. The emergence of this new class of myopathies underscores the urgent need to develop specific therapeutic strategies that can directly target dysfunctional satellite cells. Significant progress has been made in the development of cell therapies, gene therapies, advanced delivery systems, small-molecule treatments, and physical exercise interventions aimed at restoring satellite cell function and enhancing muscle regeneration.
Despite these advances, several major challenges remain. Efficient and selective delivery of therapeutic agents to mitotically quiescent satellite cells is still limited, and our understanding of how environmental and systemic factors, such as inflammation, aging, and the muscle niche, modulate satellite cell behavior remains incomplete. Emerging technologies, including single-cell and spatial omics, will contribute in mapping the molecular heterogeneity of satellite cells in both healthy and diseased muscle, thereby revealing new therapeutic targets. A deeper understanding of satellite cell biology and its interactions with the muscle microenvironment is critical to develop precision medicine approaches.
The complex pathogenic mechanisms of satellite cell-opathies highlight the importance of combined strategies to maximize therapeutic efficacy. Future research should prioritize the integration of gene-based, pharmacological, and lifestyle interventions, supported by advanced delivery platforms such as nanoparticles and exosomes to reach the satellite cell compartment more effectively. For example, pharmacological agents or niche-targeting interventions can provide a pro-regenerative environment for satellite cells, and simultaneously optimize the efficacy of gene correction or cell-based therapies that can address the underlying genetic defects. Similarly, physical exercise and metabolic modulators can synergize with molecular therapies to improve muscle repair and function. By leveraging complementary mechanisms, combined therapies may overcome the limitations of single approaches, providing more durable and comprehensive restoration of muscle regenerative capacity, which will be crucial to move from symptomatic management toward curative interventions.
Footnotes
Acknowledgements
P.G. is supported by fellowships from Neuromuscular Disease Network for Canada (NMD4C)/Muscular Dystrophy Canada (MDC) and the Canadian Institutes of Health Research (CIHR). I.M. is supported by an FRQS (Fonds de Recherche du Québec- Santé) scholarship. N.AD is supported by a Canada Research Chair and by research grants from CIHR. We acknowledge the support of the Quebec Cell, Tissue and Gene Therapy Network –ThéCell (a thematic network supported by the FRQS).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Canada Research Chairs, (grant number CRC-2024-00025), NMD4C/MDC, FQRS, and CIHR.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.