
Abstract
Background
Dynactin 1 (DCTN1) mutations are associated with diverse neurological disorders, including distal hereditary motor neuropathy, Perry syndrome, and amyotrophic lateral sclerosis. This study focused on a family with symptoms resembling spinal and bulbar muscular atrophy, showing severe vocal cord paralysis, to understand DCTN1-related neurological disorders in Koreans.
Method
Clinical examinations revealed variable phenotypes, such as proximal limb weakness, chronic hypercapnia, and gynecomastia, alongside vocal cord paralysis. Whole-exome sequencing identified a missense mutation, c.1175G > A, in DCTN1. Three more Korean families with the same mutation were analyzed to explore a potential founder effect. Microsatellite analysis indicated a shared haplotype, suggesting a common genetic origin.
Result
This study identified a missense mutation, c.1175G > A, in DCTN1 in the initial family with features resembling spinal and bulbar muscular atrophy. The mutation was also present in three other Korean families, indicating a potential founder effect. Microsatellite analysis confirmed a shared haplotype among these families. Meanwhile, the patients also manifested additional clinical features such as peripheral neuropathy and gynecomastia.
Conclusion
This study highlights clinical heterogeneity in Korean patients with DCTN1-associated neurological disorders and identifies a potential founder mutation, c.1175G > A, expanding the clinical spectrum of DCTN1 mutations with clinical features of spinal bulbar muscular atrophy. Understanding such genetic and clinical diversity is crucial for accurate diagnoses and management, with implications for future research and therapeutic strategies.
Introduction
DCTN1 is a protein that plays an important role in the interaction with microtubules and the stabilization of the neuronal cytoskeleton. Mutations in the DCTN1 gene cause axonal damage and motor degeneration. Moreover, DCTN1 mutations are known to cause variable phenotypes in neurological diseases encompassing distal hereditary motor neuropathy (dHMN); meanwhile, DCTN1 mutations can manifest as various clinical features, such as Perry syndrome and amyotrophic lateral sclerosis (ALS). Studies have tried to classify these clinical features according to onset age and the locations of distal limb weakness and atrophy in early adulthood; meanwhile, symptoms such as parkinsonism, frontotemporal dementia, and central hypoventilation occur in later ages. Other studies have attempted to classify the features according to the mutation sites to expand the clinical and genetic features of DCTN1-related spectrum diseases, i.e., the ALS-related symptoms are likely linked to the C-terminal region; meanwhile, Perry syndrome was related to the CAP–Gly domain.
Here, we report on a family eventually diagnosed with DCTN1 that exhibited an additional phenotype mimicking symptoms similar to spinal bulbar muscular atrophy, e.g., severe vocal cord paralysis in the early stage of the disease alongside prominent gynecomastia. We further analyzed three other DCTN1 variants within different families in Korea to determine the genotype–phenotype correlation.
Materials and methods
Participants
Patients from four different families were included in this study. The KNUCH-F01 was studied extensively with two affected and five unaffected family members. This study also included one patient from three separate families with identical and different mutation sites.
Whole-exome sequencing
Blood samples were collected from one patient and family members, and the genomic DNA was extracted using the FlexiGene DNA kit (Qiagen, Hilden, Germany). We used 10 μg of genomic DNA to construct the library using a SureSelect V6-post kit (Agilent Technologies, Santa Clara, CA, USA). We sequenced the captured DNA fragments using HiSeq 4000 (Illumina, San Diego, CA, USA). Data were mapped to the reference human genome, hg19, from the University of California, Santa Cruz (GRCh37 from NCBI, Feb. 2009) using BWA (Ver. Bwa-0.7.12), Picard (Ver. Picard-tools-1.130), GATK (Ver. GATKv3.4.0), and SnpEff (Ver. SnpEff_v4.1 g). The genetic variants were annotated using databases, including the 1000 genome release phase 3 (http://www.internationalgenome.org), dbSNP build 142, and ClinVar release 05/2015 from National Center for Biotechnology Information (NCBI, httP://www.ncbi.nlm.nih.gov), and NHLBI Exome Sequencing Project (ESP, release ESP6500SI_V2, http://evs.gs.washington.edu/EVS/).
Sanger sequencing
Exon 2 and the exon–intron boundaries of the DCTN1 gene were amplified by polymerase chain reaction (PCR) using h-Taq polymerase (Solgent, Daejeon, Korea). Specific primers were designed using Primer3 v0.4.0 (http://bioinfo.ut.ee/primer3-0.4.0/). The PCR products were digested using shrimp alkaline phosphatase (USB, Cleveland, OH, USA) and exonuclease I (USB) to remove unincorporated nucleotides and primers. The purified PCR products were sequenced using the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) and the 3130xl genetic analyzer (Applied Biosystems). Sequencing data were analyzed using Chromas Pro v1.5 (Technelysium Pty Ltd, Tewantin, QLD, Australia). Sequencing data from the patients were compared with reference DCTN1 gene sequences (NM_004082.4) obtained from the NCBI database (http://blast.ncbi.nlm.nih.gov).
Microsatellites analysis
Chromosome 2, which houses the DCTN1 gene, was designated the primary target for microsatellite analysis. Primer sequences were meticulously designed to facilitate this analysis, incorporating a fluorescent FAM tag at the 5′ end. PCR amplification was performed using E-Taq polymerase (Solgent, Daejeon, Korea). The specific primer sequences were designed using Primer 3 v0.4.0 (http://bioinfo.ut.ee/primer3-0.4.0/). Subsequently, the resulting amplified products were subjected to sequencing using the 3130xl genetic analyzer (Applied Biosystems). Microsatellite data were analyzed using GeneMapper Software 5 (Applied Biosystems).
Results
Clinical findings
The proband (II-8) in the family KNUCH-F01 was a 58-year-old male who presented with proximal limb weakness and dyspnea that had slowly progressed for 20 years (Figure 1). The patient complained of difficulty climbing, orofacial fasciculation, and a hand tremor at 37 years old. At the age of 45, the patient experienced chronic fatigue and mild dyspnea and later developed severe dysphonia. The patient was diagnosed with bilateral vocal cord palsy and hypercapnia that became aggravated during sleep. The patient also noticed gynecomastia and, at the age of 53, experienced recurrent loss of consciousness due to severe hypercapnia. The initial neurological examination of the patient showed moderate dysarthria with prominent orofacial fasciculation. The patient also had tongue atrophy and spontaneous fasciculations, while the rest of the cranial examination was unremarkable. The patient had asymmetric bilateral first dorsal interosseous muscle atrophy and presented a Medical Research Council (MRC) rating of 4 in the proximal lower limbs. The deep tendon reflexes were absent without pathologic reflexes, and the patient complained of distal dominant paresthesia.
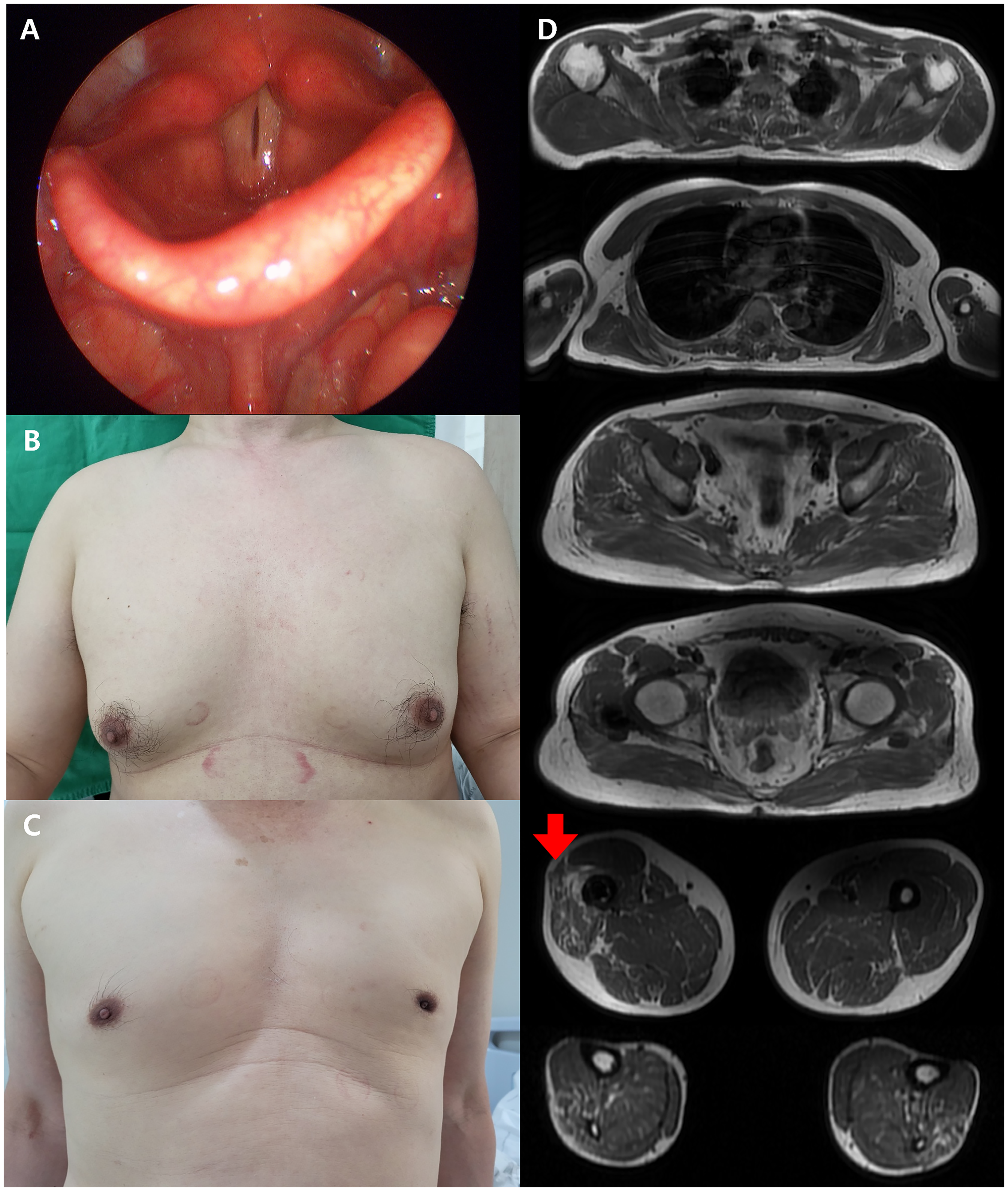
The additional phenotypes and magnetic resonance imaging of the muscles in the patients.
The laboratory tests showed a mild elevation in creatine kinase (CK) (383 IU/L) levels, while the thyroid function tests and hormonal levels, including luteinizing hormone, follicular stimulating hormone, and testosterone, were within normal ranges. The arterial blood gas analysis showed mild hypercapnia with a CO2 level of 58.8. The mini-mental status examination (MMSE) score was 29, and the global depression scale (GDS) score was 8. An electrophysiological study showed mild peripheral neuropathy, and the needle electromyography study exhibited diffuse giant motor unit potentials in all the tested muscles. The magnetic resonance imaging (MRI) revealed mild fatty infiltration in the thigh and calf muscles, suggesting a neuromuscular disease. The pulmonary function test showed a decreased forced vital capacity of 58%. The tentative clinical diagnosis suggested complex phenotypes of peripheral neuropathy, spinal and bulbar muscular atrophy (Kennedy disease), however, the CAG repeats in the androgen receptor (AR) gene were normal. The nephew of the patient presented similar symptoms, suggesting a family history.
The nephew (III-3) who exhibited similar symptoms was a 42-year-old male (Figure 1). The nephew experienced mild dysarthria at the age of 39 years. At the age of 41, the nephew complained of orofacial fasciculation and cramps. The nephew began to experience dysphagia and hypercapnia that caused him to awaken during sleep; thus, continuous positive airway pressure was employed at night. The initial neurological examination showed moderate dysarthria with orofacial fasciculation and tongue atrophy. The cranial nerves were intact; however, there was evidence of bilateral vocal cord paralysis. The motor function test revealed mild proximal leg weakness with an MRC grade of 4. The deep tendon reflexes were decreased, and bilateral gynecomastia was noted. The laboratory test showed an increased CK level of 619 IU/L; other tests, including thyroid, liver, and renal function, were within normal ranges. The initial arterial blood gas analysis showed an increase in pCO2 to 55.4.
The nerve conduction study showed mild sensory dominant peripheral neuropathy, and the needle electromyography showed high amplitude MUPs. The pulmonary function test presented an FVC of 88%. The cognitive function test revealed an MMSE of 30 and a GDS score of 6. An MRI of the muscles of the III-3 patient revealed fatty infiltration in the bilateral thigh.
Notably, two additional patients who were family members died suddenly, aged in their 40s, and all four patients experienced waking up in the night due to dyspnea, potentially reflecting chronic hypercapnia. Moreover, both the proband and the nephew had clinical gynecomastia. Both patients had normal hormonal levels (estrogen and progesterone) and liver and renal function tests. The patients had no clinical history of taking hormonal drugs such as steroids, spironolactone, digoxin, calcium channel blockers, psychogenic drugs, or alcohol, which could influence iatrogenic gynecomastia.
We further analyzed three patients from different families with putative DCTN1 pathogenic mutations (Table 1). These second and third families were previously described in our existing literature and were included; 1 a fourth patient, newly diagnosed with sensorimotor neuropathy, was also added.
Clinical and electrophysiological features in Korean patients with the DCTN1 mutations.
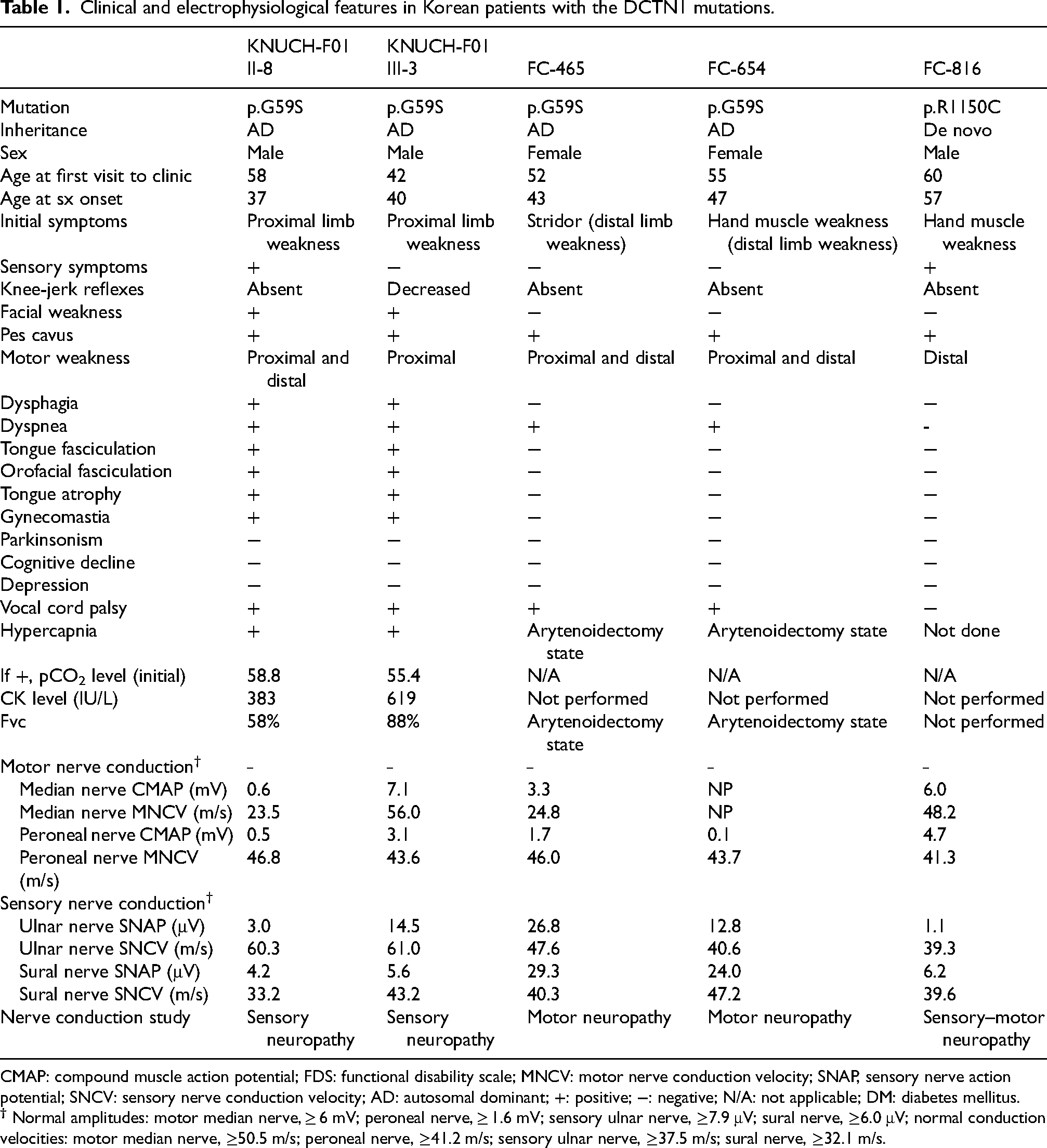
CMAP: compound muscle action potential; FDS: functional disability scale; MNCV: motor nerve conduction velocity; SNAP, sensory nerve action potential; SNCV: sensory nerve conduction velocity; AD: autosomal dominant; +: positive; −: negative; N/A: not applicable; DM: diabetes mellitus.
Mutation analysis
Whole-exome sequencing of the affected (Ⅱ-7, II-8, Ⅲ-3) and unaffected (Ⅱ-5, Ⅱ-6, Ⅲ-1, Ⅲ-2) family members with an autosomal dominant inheritance pattern was performed (Figure 2A). A 60.4 Mbp area in the target region was identified, with ≥96% of the bases covered with a depth >10× (Table 2). In total, 174,196 variants were identified in the seven family members. To identify pathogenic variants, we filtered out variants in non-coding regions, synonymous variants, and variants with allele frequencies <0.01 using the 1000 Genomes, dbSNP, ClinVar, and ESP databases. Only one variant remained in the family analysis of the remaining variants, denoting dominant inheritance (Figure 2B). Finally, a missense mutation was identified in DCTN1, c.1175G > A (p.G59S), an associated motor neuron disease gene. 2
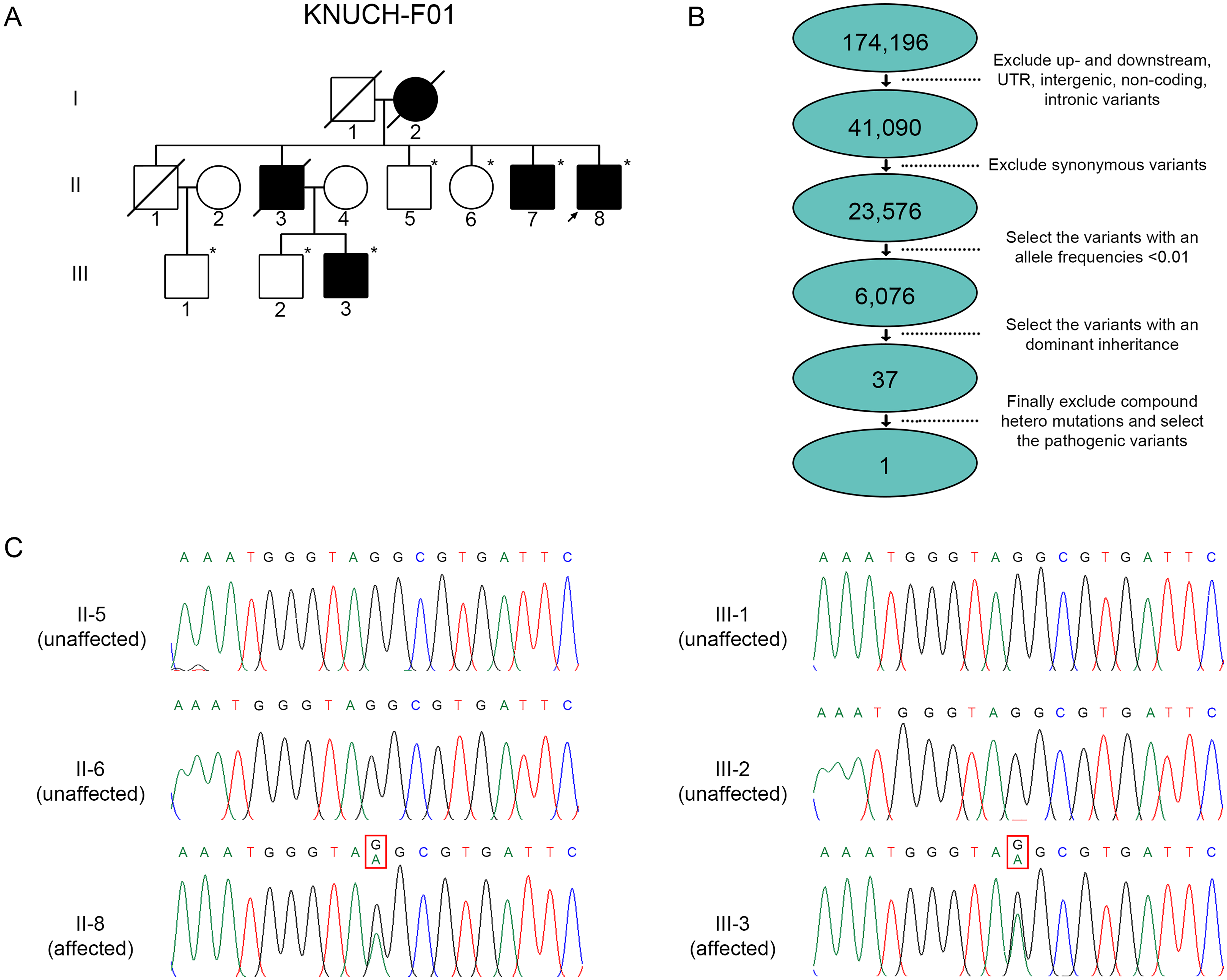
Analysis of pedigrees with the p.G59S mutation in DCTN1.
Run statistics and coverage of whole-exome sequencing.

In addition to KNUCH-F01, the same mutation was confirmed in two other Korean families. This mutation, previously identified in FC-465 and FC-654, was analyzed in these two additional Korean families using identical Sanger sequencing techniques. 1 This variant was confirmed by Sanger sequencing, and the mutation was co-segregated with the affected individuals (Figure 2C).
To investigate the effect of a founder mutation in familial settings, we conducted a gene analysis of patients and healthy individuals from three Korean families associated with the DCTN1 gene. We employed a sequencing analysis using a total of 18 genetic markers. Among these markers, we focused on the region surrounding the p.G59S mutation using two single nucleotide polymorphism (SNP) markers, while regions further away were analyzed using 16 short tandem repeat (STR) markers. In cases where the microsatellite analysis presented inconclusive results, the corresponding data were excluded from the study. The distance between the 5′-STRs8 marker and the 3′-STRs8 marker was approximately 120 Mbp (Figure 3). The markers for genetic analysis were selected by referencing databases, such as the UCSC's dbSNP155, dbSNP Archive, RepeatMasker, Simple Repeats, and Microsatellite database.
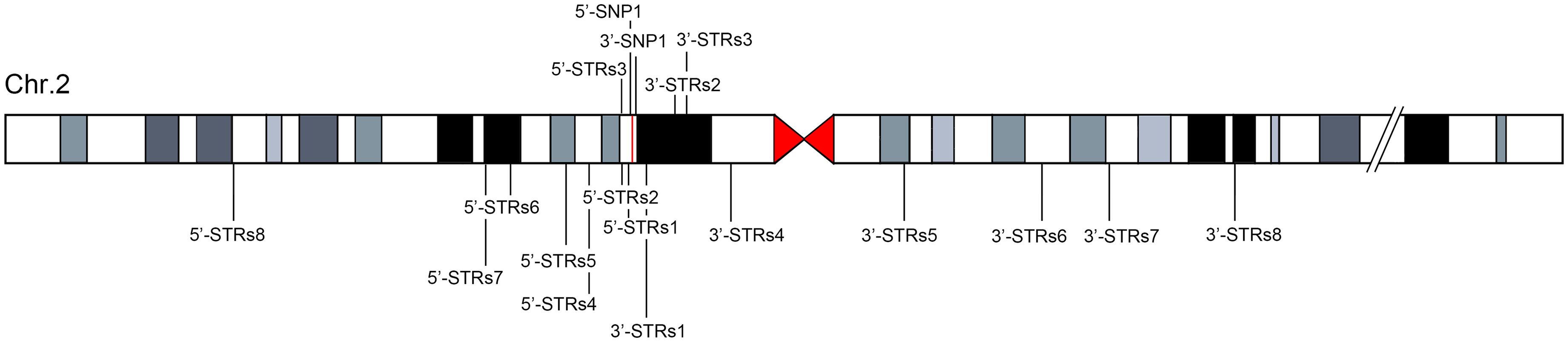
Location of the p.G59S mutation in chromosome 2 and linked SNP and STR markers.
For the fragment analysis of STRs, we examined fluorescent peaks and identified heterozygous peaks based on the repeat number of STRs (Table 3). Consistent with the sequencing results for the DCTN1 gene mentioned earlier, the patients (KNUCH-F01: II-8, III-3; FC-465; FC-654) exhibited heterozygosity within Chromosome 2, with the c.1175G > A mutation resulting in a heterozygous A and G base sequence. In contrast, a fragment analysis was performed to compare and analyze FC-816, which possessed the R1150C mutation instead of the G59S mutation. Subsequent analysis of the 5′-SNPs1 and 3′-SNPs1 markers revealed identical T and G base sequences, respectively, in the patients.
Microsatellite analysis of subjects carrying p.G59S and their family members.
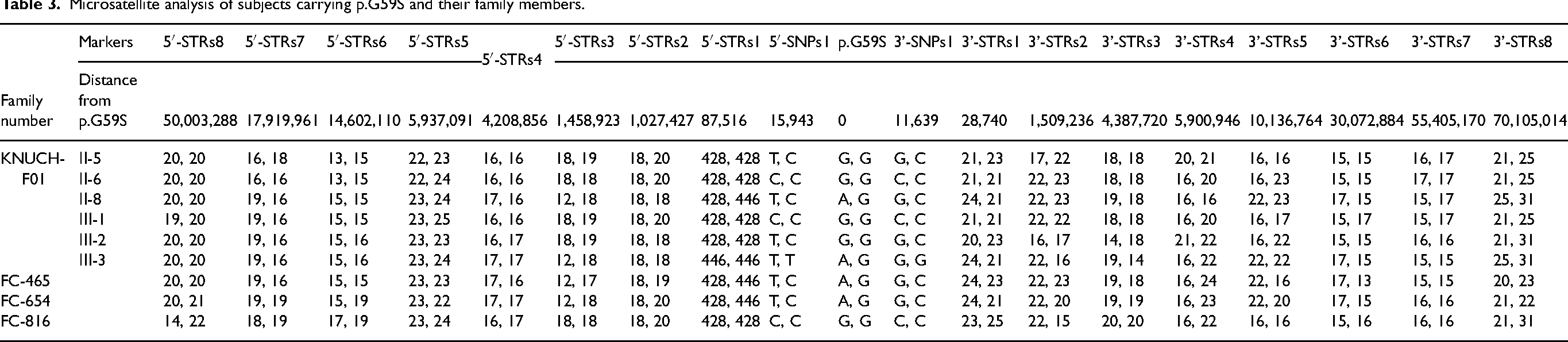
The SNP analysis estimated a shared haplotype spanning approximately an 80 Mbp region from 5′-STRs8 to 3′-STRs6 in the KNUCH-F01, FC-465, and FC-654 patients. However, this haplotype was not observed in the FC-816 patient, who possessed a different mutation from the other families. This finding suggests that the p.G59S variant, found at a relatively high frequency in Koreans, may have originated from a common founder (Table 3).
Discussion
Clinically, the DCTN1 mutation can promote various neurodegenerative diseases, including dHMN, Perry syndrome, and amyotrophic lateral sclerosis. 3 A recent study demonstrated the existence of a mutation site in the CAP–Gly domain of DCTN1 that may affect the clinical phenotype of Perry syndrome, which typically shows Parkinsonism. Meanwhile, the ALS- and dHMN-related mutations are mostly harbored in the non-CAP–Gly domain or the C-terminal region in the DCTN1 gene. Notably, the DCTN1-related Perry syndrome, which manifests with Parkinsonism and exhibits levodopa-responsive bradykinesia, resting tremor, rigidity, and postural instability, can lead to a misdiagnosis of Parkinson`s disease in the early disease stages. Indeed, two of our newly diagnosed patients presented clinical features including orofacial fasciculation, gynecomastia, and peripheral neuropathy. Interestingly, none of our patients presented clinical features of Parkinsonism or clinically significant depression, which is normally observed in Perry syndrome; however, our patients harbored a G59S mutation in the CAP–Gly domain that differs from previous studies. Moreover, all of our patients showed proximal limb weakness with prominent bilateral vocal cord palsy that led to arytenoidectomy; these symptoms are in accordance with the identical mutation described previously. 1 Our patients showed additional clinical features encompassing chronic hypercapnia and gynecomastia with orofacial fasciculation and weakness in the early stage of the disease. These additional clinical findings are cardinal features observed in spinal and bulbar muscular atrophy, yet our patients possessed normal CAG repeats in exon 1 of the AR gene.
Among these noted additional features, gynecomastia in males can be caused by various processes; gynecomastia is a benign enlargement of breast glandular tissue in males due to an imbalance in estrogen and androgens. Therefore, systemic diseases such as liver and kidney disease should be assessed along with hormonal abnormalities. Iatrogenic gynecomastia should also be considered, and studies related to the consumption of hormonal drugs, spironolactone, digoxin, calcium channel blockers, psychoactive drugs, cimetidine, ketoconazole, and alcohol need to be checked.
Our patients were relatively young or middle-aged but had no underlying liver or kidney disease. The patients had no past medical history that could influence iatrogenic gynecomastia, leading to idiopathic gynecomastia, which is frequently observed in SBMA. Notably, our patients presented with clinical features of SBMA with hypercapnia. A recent study showed that 3 out of 16 SBMA patients had hypercapnia without significant clinical symptoms. 4 Therefore, although rare, hypercapnia is always possible, even in SBMA, and needs to be considered during the diagnosis.
A recent study reported differences in the non-CAP–Gly domain that presented a typical DCTN1-related neuropathy with additional clinical features, such as congenital foot deformity. This observation differed from our patients, who still showed clinically significant clinical features, but following sequencing differences in the CAP–Gly domain, presenting a high prevalence of Parkinsonism and other symptoms that involve the central nervous system; these findings expand the clinical spectrum of DCTN1-related disease.
This study conducted a further gene analysis for our patient group and healthy individuals from two Korean families, along with the identical mutation site reported in a previous study, to investigate the effect of a founder mutation in familial settings. We also added a newly diagnosed patient from a different family (FC-816). Our analysis focused on the DCTN1 gene and employed a sequencing analysis using 18 markers. To narrow our analysis, we specifically focused on regions within 20,000 bp of the p.G59S reference using three SNP markers. For regions further away, we utilized 16 STR markers. In cases where the microsatellite analysis yielded inconclusive results, the corresponding data were excluded from the analysis.
The G59S mutation in the DCTN1 gene is located within the CAP–Gly domain. The CAP–Gly domain primarily binds microtubule-related proteins and regulates their functions and interactions. 5 Therefore, this domain plays a critical role in controlling the dynamic properties of microtubules. The CAP–Gly domain also modulates interactions between the dynactin complex and other proteins or cellular components, contributing to diverse cellular processes. 6 Previous research findings have indicated that a significant number of mutations in the DCTN1 gene occur within this domain, indicating that this region serves as a genetic hotspot (Figure 4).7–19
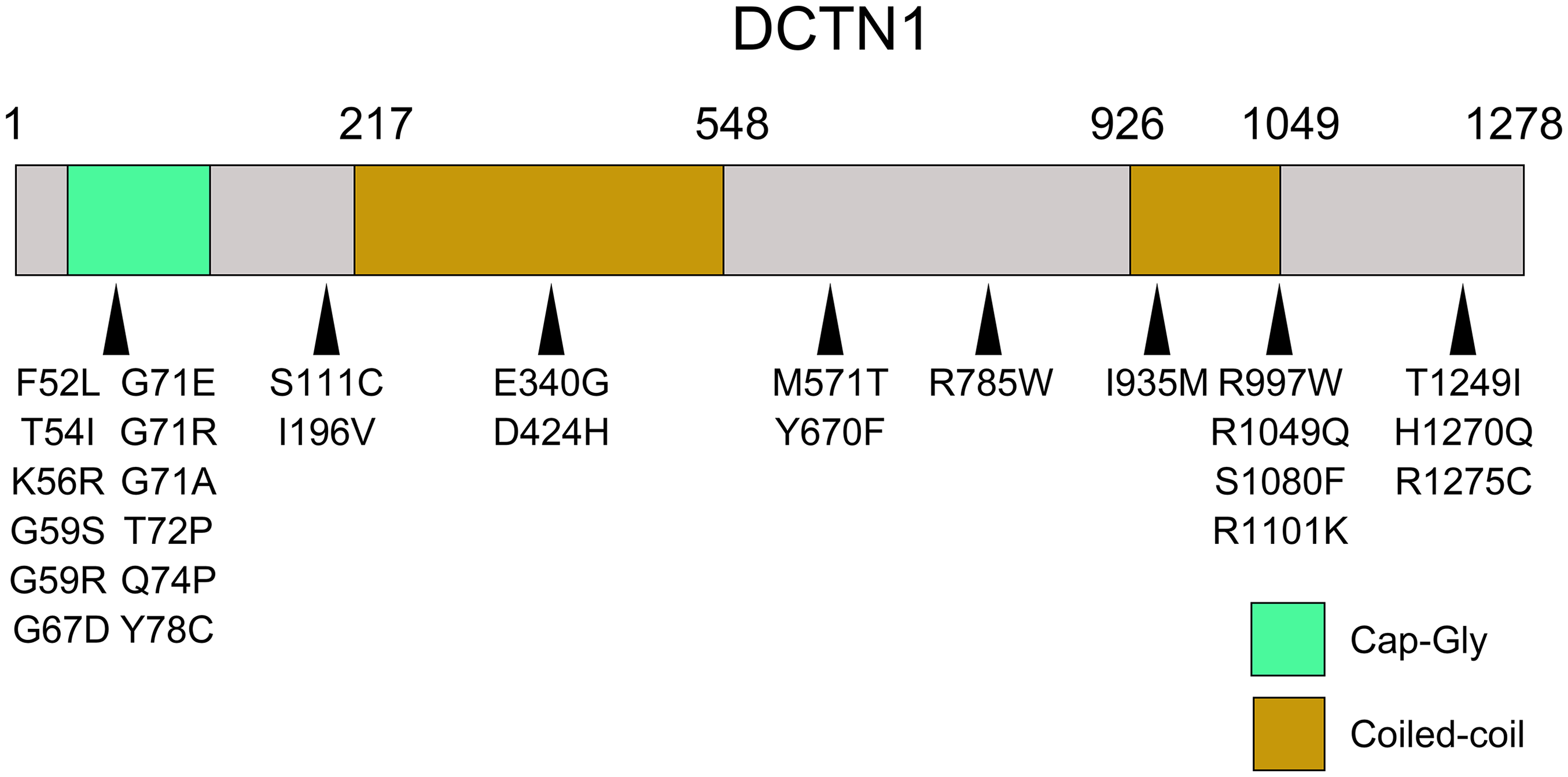
Schema of the DCTN1 mutations.
Haplotype analysis revealed that the KNUCH, FC-465, and FC-654 families all harbored the G59S mutation and displayed an identical number of microsatellite repeats between 5′-STRs8 and 3′-STRs6, encompassing a DNA sequence of approximately 100 Mbp. This finding indicates a significant association between various genomic regions and the G59S mutation. Notably, despite the lack of familial relatedness among the KNUCH, FC-465, and FC-654 families, the shared identical microsatellite repeat numbers over a considerable genomic distance suggest a likely inheritance pattern from a common ancestor. Thus, these results provide compelling evidence for a shared genetic origin of the G59S mutation within these families. In support of this, additional evidence stems from FC-816 individuals carrying the R1150C mutation instead of G59S. Interestingly, unlike the above families, these individuals displayed consistent microsatellite repeat numbers within a relatively shorter segment, specifically from 5′-STRs4 to 3′-STRs1. This observation implies that the shared ancestry associated with the G59S mutation is not a general characteristic among the entire Korean population but rather linked to the specific context of the G59S mutation.
This clinical aspect also correlates with the above genetic findings. Despite the same G59S mutation, the KNUCH family clinically presented with proximal limb weakness, orofacial fasciculation, and gynecomastia. The patient also had sensory symptoms that were clinically compatible with spinal bulbar muscular atrophy. However, the KNUCH families underwent genetic studies to exclude a diagnosis of SBMA, and the CAG repeats in the AR gene were within the normal ranges. Conversely, the identical G59S mutation in families 2 and 3 promoted a distal dominant weakness without sensory symptoms, and these patients showed no clinical features, including orofacial fasciculation or gynecomastia. Notably, family four, which harbored a different R1150C mutation, showed sensory symptoms that correlated with the nerve conduction study, in accordance with KNUCH families. Moreover, the patient from family four manifested distal dominant weakness with sensory symptoms combined with postural tremor that were clinically suggestive of Charcot-Marie-Tooth disease type 2, which bridges the clinical symptoms observed in other G59S harboring patients. Our clinical findings reflect that the DCTN1-related disease can show variable phenotypes despite the same mutation site; thus, further studies are needed to understand the underlying pathophysiology.
In conclusion, our study provides valuable insights into the effect of a founder mutation in South Korea. By utilizing sequencing analysis and fragment analysis of STRs, we were able to identify and characterize the presence of specific mutations in the DCTN1 gene in different Korean families. The observed conservation of base sequence movement within each family and across pedigrees highlights the potential significance of this founder mutation in the pathogenesis of the disease. Moreover, we expanded the clinical spectrum of the G59S mutation in DCTN1, which presented with proximal limb weakness, chronic hypercapnia, and gynecomastia—features typically observed in SBMA patients. These additional findings broaden the clinical phenotype of DCTN1, and these additional clinical symptoms should be considered as a phenotype in DCTN1-related neuropathies. Lastly, our study evaluated the founder effect that may contribute to further understanding the genetic basis of familial diseases and may have implications for genetic counseling and future research.
Footnotes
Ethics considerations
This study was approved by the Institutional Review Board of Kyungpook National University Chilgok Hospital (KNUCH 2019-11-012), which was conducted in conformity with the Declaration of Helsinki.
Author contributions
BL: Acquisition of data, analysis and interpretation of data, drafting and revising the manuscript. STC: Acquisition of data, analysis and interpretation of data, drafting the manuscript. YRK: Acquisition of data, analysis and interpretation of data, drafting the manuscript. RK, KWC, TJK: Acquisition of data, analysis and interpretation of data. BOC: Conceptualization and design of the study, revising the manuscript. UKK: Conceptualization and design of the study, revising the manuscript, analysis and interpretation of data, Acquisition of funding. JSP: Conceptualization and design of the study, drafting and revising the manuscript, analysis and interpretation of data, Acquisition of funding.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Research Foundation of Korea (NRF), which is funded by the Korean government [Ministry of Science and ICT; 2022R1A2C3003700 (to UKK.), 2022R1F1A1068420 (to B.L) and 2022R1C1C1009723 (to J.S.P.)], by the Korean Fund for Regenerative Medicine [Ministry of Science and ICT, and Ministry of Health and Welfare; 23C0115L1 (to B.O.C.)], and by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) [Ministry of Health and Welfare; HR22C1363 (to B.O.C.)].
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.