
Abstract
Pathogenic variants in the Glycyl-tRNA synthetase gene cause the allelic disorders Charcot-Marie-Tooth disease type 2D and distal hereditary motor neuropathy type V. We describe clinical features in 8 unrelated patients found to have Glycyl-tRNA synthetase variants by Next Generation Sequencing. In addition to upper limb predominant symptoms, other presentations included failure to thrive, feeding difficulties and lower limb dominant symptoms. Variability in the age at testing ranged from 14 months to 59 years. The youngest being symptomatic from 3 months and ventilator-dependent. Sequence variants were reported as pathogenic, p.(Glu125Lys), p.(His472Arg); likely pathogenic, p.(His216Arg), p.(Gly327Arg), p.(Lys510Gln), p.(Met555Val); and of uncertain significance, p.(Arg27Pro). Our case series describes novel Glycyl-tRNA synthetase variants and demonstrates the clinical utility of Next Generation Sequencing testing for patients with hereditary neuropathy. Identification of novel variants by Next Generation Sequencing illustrates that there exists a wide spectrum of clinical features and supports the newer simplified classification of neuropathies.
Keywords
INTRODUCTION
Charcot-Marie-Tooth (CMT) is a clinically and genetically heterogeneous group of disorders with more than 80 genes associated with different subtypes [1]. Two of these subtypes include CMT type 2D (CMT2D) and distal hereditary motor neuropathy type V (HMNV), which are considered to be allelic disorders. Typically, these conditions are characterised by adolescent or early adult-onset hand and upper limb-predominant weakness and wasting, although lower limb involvement is also seen in approximately half of patients. It has traditionally been considered that the two diseases can be differentiated clinically, with only CMT2D patients developing sensory deficits. Although originally considered to be distinct diseases, independent family studies mapped these phenotypes to a region on chromosome 7p suggesting they were allelic [2]. This region was later defined by molecular testing and the gene responsible was identified as Glycyl-tRNA synthetase (GARS) [3].
GARS belongs to the family of aminoacyl-tRNA synthetase enzymes, which perform essential functions in protein synthesis by charging tRNA molecules with amino acids. In this way, the protein encoded by GARS is responsible for catalysing the ligation of glycine to the 3’-end of its cognate tRNA. Several pathogenic autosomal dominant variants in GARS have been found to confer reduced aminoacyl-tRNA synthetase activity [4]. Using results from our targeted 56-gene Next Generation Sequencing (NGS) panel assay we collate clinical information, and where available neurophysiology results, from patients identified with GARS variants.
MATERIALS AND METHODS
The Bristol Genetics Laboratory, UK provides genetic analysis for NHS patients with inherited and acquired disorders. The laboratory is UKAS-accredited and offers a range of UK Genetic Testing Network (UKGTN)-approved services, including an NGS service for the genetic diagnosis of peripheral neuropathies. This is a custom-designed SureSelect (Agilent Technologies) solution-based oligonucleotide target capture assay consisting of 56 genes [5]. Since the test was established in 2013, the laboratory has received approximately 2000 diagnostic requests from regional, national and international users and has a positive detection rate of approximately 35% [5]. A review of 1818 patients tested on this gene panel identified a total of 21 patients with candidate disease-causing GARS variants. This provides a diagnostic yield for the GARS gene of up to 1.2% in our cohort.
Out of the 21 patients identified in our cohort, 7 families signed a declaration consenting for their medical data to be included in this publication. Responsible clinicians provided clinical details for all patients, and 3 provided neurophysiological data. One additional patient tested in an Italian genetics laboratory using a similar approach, has also consented to be included in this report (Patient 2). All the patients in this series are reported anonymously, except to their treating clinician. Specific ethical approval was sought from the local clinical ethics committee but deemed to not be necessary.
RESULTS
Clinical findings
The clinical features of all 8 patients are listed in Table 1. The 3 patients whose neurophysiological assessments were available are listed in Table 2. Most patients had both upper and lower limb involvement, however it was not ascertained if their symptoms were always more severe in the upper limbs as typically described in CMT2D and HMNV. There was only one reference to distal sensory loss and slightly reduced sensory nerve action potential, suggesting a diagnosis of CMT2D (Patient 8). The remaining 7 patients, who appeared not to display any sensory symptoms, were suggested to have phenotypes consistent with a diagnosis of HMNV. In accordance with either diagnosis, common clinical features in our cohort included foot deformities, distal weakness and wasting. Variability in the age at testing ranged from 14 months to 59 years.
Details of all 8 patients included in this cohort, including their age of onset, clinical features and family history and GARS variants identified through genetic testing
Nerve conduction study and needle electromyography results from 3 of the patients included in this cohort
Abbreviations: CMAP, compound muscle action potential; DML, distal motor latency; EMG, electromyography; MCV, motor conduction velocity; NCS, nerve conduction study; SNAP, sensory nerve action potential; LL, lower limb; UL, upper limb.
Patient 1 was the oldest patient tested in our cohort and developed symptoms in his hands from the age of 30 years. The weakness was predominantly in the thenar and hypothenar muscles and had been previously attributed to vibratory damage due to his occupation involving the use of a pneumatic drill. By the age of 50 years he also developed symptoms in his lower limbs. His past medical history was of a right-sided parietal subdural haematoma evacuated by a craniotomy. He had slurred speech, complained of a feeling of ‘mugginess’ in his head and allegedly had diplopia which was not confirmed on clinical examination. A strong candidate GARS variant of uncertain significance, p.(Arg27Pro), was detected in this patient and also in his similarly affected daughter with distal arm weakness and wasting.
Patient 2 harboured a previously reported pathogenic GARS variant, p.(Glu125Lys), and was the youngest and most severely affected patient with widespread neurogenic changes, being symptomatic from 3 months. The clinical presentation in this patient included failure to thrive and severe muscle weakness which was slightly more marked in lower limbs and distally. This infant was ventilator-dependent from age 7 months and also had feeding difficulties leading to a percutaneous gastrostomy with fundoplication.
Patient 5 was a young male who was prone to falling over but reported no obvious weakness or other physical symptoms. Clinical examination revealed his tone, co-ordination and muscle power to be normal except for ankle dorsiflexors which were mildly weak. Ankle jerks were depressed and there was evidence of hammer toes and pes cavus. Clinical testing of the hands also revealed weakness of the thenar eminence and muscles surrounding the thumb (Fig. 2A-B). This patient’s father was reportedly symptomatic from 14 years, and upon examination was found to have profound hand wasting of the intrinsic muscle and first dorsal interossei resulting in a clawing. His lower limbs were wasted with considerable loss of muscle bulk around the calves and he required crutches to ambulate. Additionally, he was affected with severe weakness of dorsiflexion, profound pes cavus and hammer toes. The patient’s paternal grandmother was also examined and had mild hand and wrist weakness with slight wasting of the thenar eminence, but otherwise she was asymptomatic. Extensive molecular genetic studies were previously carried out on this heterogeneous family, including sequential single gene sequencing of neuropathy-related genes, all of which delivered uninformative results. Finally the application of large gene panel NGS testing led to the identification of the well characterised p.(His472Arg) GARS variant (Fig. 2C). While this variant segregated with disease in the family and was deemed in keeping with the clinical features described, there were no other candidate variants detected in other genes to help explain the heterogeneity across the family.
Genetic findings
A total of 7 unique GARS variants were identified by NGS and confirmed by Sanger sequencing (Table 3). These were all missense changes akin to the majority of pathogenic GARS variant types described in the literature. Consistent with previous reports, these variants were distributed throughout the gene, and not restricted to a particular hot-spot or functional protein domain (Fig. 1).
Details of the GARS variants identified in this cohort through genetic testing, including their reported and ACMG classifications. Sequence variant nomenclature based on transcript NM_002047.2
Abbreviations: ACMG, American College of Medical Genetics and Genomics.
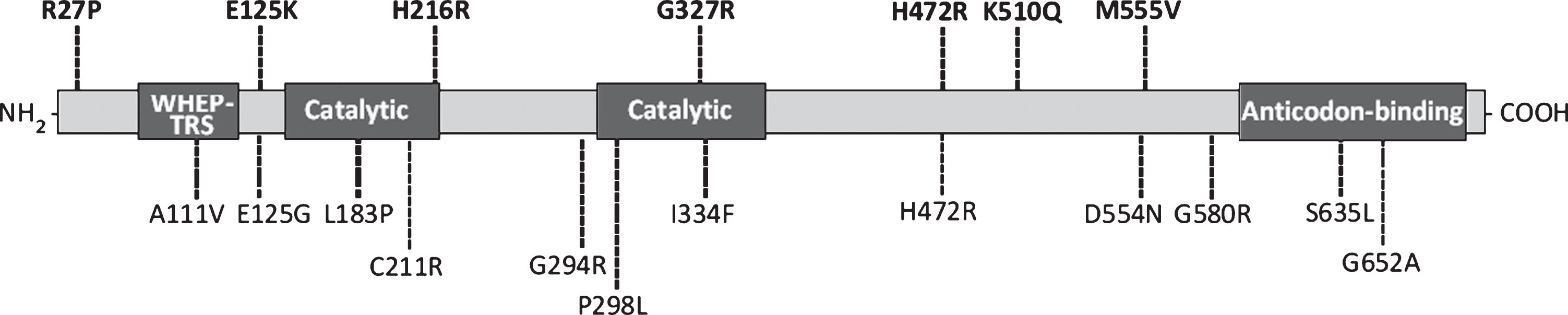
Schematic presentation of the GARS protein and functional domain structure. Variants identified in this study are shown above the protein schematic in bold and previously reported variants are shown below it. Amino acid nomenclature based on transcript NM_002047.2 (739 a.a).
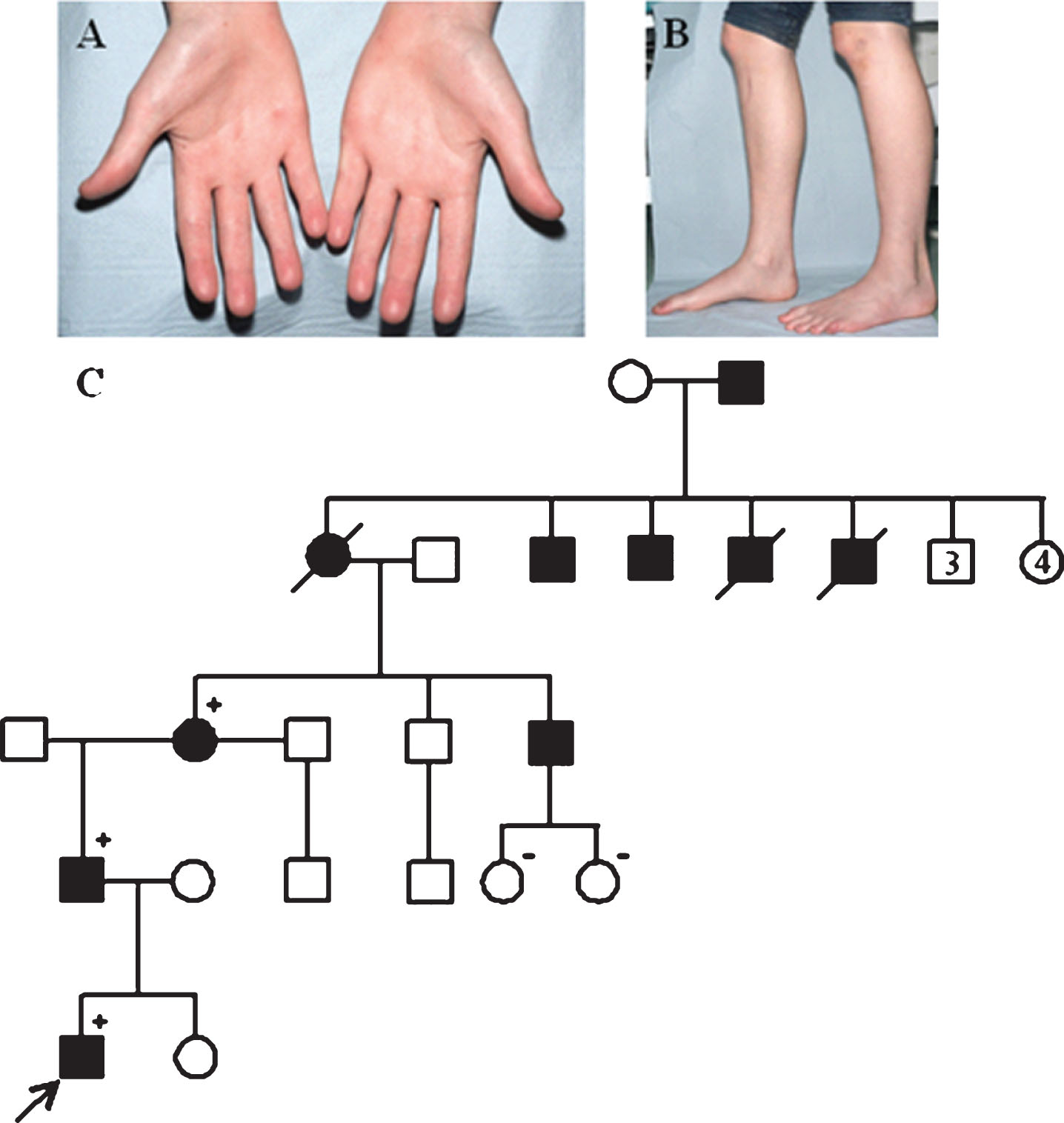
Weakness of the thenar eminence and muscles surrounding the thumb (A) and feet (B) in Patient 5. Segregation of the c.1415A>G, p.(His472Arg) GARS variant. The proband (Patient 5) is indicated by the arrow. Squares represent males and circles females. Filled symbols represent individuals with a clinical diagnosis of peripheral neuropathy, open symbols represent unaffecteds. Plus signs and minus signs depict variant-positive and variant-negative individuals, respectively. Crossed symbols represent deceased individuals (C).
At the time of reporting to patients from the two genetic laboratories, the pathogenicity and clinical significance of variants were assessed using a combination of in silico analysis tools, literature searches and population frequency data. For the purposes of this publication, the Bristol Genetics Laboratory have now subjected the variants to classification according to the American College of Medical Genetics and Genomics (ACMG) and Association for Clinical Genomic Science (ACGS) variant interpretation best practice guidelines [6, 7]. Based on these new guidelines, several of the variants have been downgraded to a less pathogenic classification compared to when they were originally reported, however they are still considered strong candidates for the basis of disease and further investigation is required.
Four variants have not been reported in the literature before this study, to our knowledge, and therefore represent novel findings; p.(Arg27Pro), p.(Gly327Arg), p.(Lys510Gln) and p.(Met555Val). Segregation studies were performed for the novel variants p.(Arg27Pro) and p.(Lys510Gln) identified in Patients 1 and 7, respectively. Both of these variants were found to segregate with other affected relatives which provided additional evidence supporting their pathogenicity. Patient 4, who was tested at the age of 16 years, was previously included as part of a wide data set study where no specific clinical information was presented. Since then further testing has been undertaken and the p.(Gly327Arg) variant identified in this patient was absent in both unaffected parents, suggesting that it arose as a de novo event in Patient 4. While it appears that pathogenic variants in the GARS gene are most often inherited from affected parents, there have been recent reports of novel de novo pathogenic GARS variants that are associated with either childhood-onset CMT2D or severe infantile-onset HMNV [8].
The variant p.(His472Arg) was present in 2 patients and is one of the most well-characterised pathogenic changes reported in the GARS gene. Indeed, functional studies have demonstrated that the p.(His472Arg) variant reduces aminoacyl-tRNA synthetase activity to 6.25% relative to wild-type GARS [4]. Patient 3 has previously been reported in the literature where cell lines harbouring the p.(His216Arg) variant were found to impair mitochondrial metabolism in neurons [9]. The previously reported variant p.(Glu125Lys) was also identified in one patient [10].
DISCUSSION
Aminoacyl-tRNA synthetases have important contributions to the control of angiogenesis, inflammation, tumourigenesis and other vital physiopathological processes [11]. Support for a loss-of-function mechanism underlying GARS-associated neuropathy includes work demonstrating that mutant proteins mislocalise in neuronal cells, suggesting that tRNA-charging deficits play a role in disease pathogenesis [12]. However, recent evidence favours a toxic dose-dependent gain-of-function mechanism [13]. This, it is suggested, coincides with abnormal neuromuscular junction assembly, leading to synaptic degeneration and reduced survival in Drosophila models [14]. Furthermore, it has been demonstrated that the expression of pathogenic GARS variants cause the protein to acquire a neomorphic binding activity that directly antagonises the vascular endothelial growth factor (VEGF)-neuropilin 1 (Nrp1) interactions, which is an essential signalling pathway for motor neuron survival [15]. Another study has shown aberrant binding to tropomyosin receptor kinase (Trk) receptors, which leads to subtly subverting sensory neuron differentiation and/or survival during early stages of development [16].
Our objective was to bring together clinical, genetic and neurophysiological information for a cohort of patients identified with GARS variants to investigate potential genotype-phenotype associations. The data we were able to collect provides further evidence, that despite pathogenic variants in GARS being primarily associated with adolescent or early adult-onset disease, they can also cause severe disease much earlier in the infantile stage of development. We observed a phenotypic variability much more subtle than the traditional dichotomous classification into CMT2D and HMNV. Our study may therefore provide evidence to support a simplification of the classification of neuropathies as proposed by Magy et al. [17]. Those proposals consisted of reclassification using a three step process; inheritance, phenotype and genotype. In other words, our cases of CMT2D/HMNV would become AD-CMT2D-GARS under the proposals. These proposals continue to be under discussion and have not been universally accepted. It remains to be seen whether patients or clinicians find these proposals helpful.
To our knowledge, Patient 2 presented with symptoms earlier than any previous cases reported in the literature (onset at 3 months). A similarly affected infant presenting with acute respiratory failure at 10 months, has also recently been reported to have motor axonopathy associated with a different pathogenic GARS variant [18]. The authors also detected this variant in 2 siblings having been inherited from an unaffected mosaic parent; one sibling had a neuromuscular disorder but no respiratory symptoms while the other with suffered both symptoms. Akin to Patient 5 in our study, this reinforces the notion of clinical heterogeneity within families with the same GARS variant, however currently the reason for this remains unknown.
Our study also emphasises the clinical utility of testing patients with a suspected hereditary neuropathy on multi-gene NGS panels, as opposed to the historical approach of sequential gene testing. Genetic changes in GARS are only estimated to account for 3% of neuropathy diagnoses, therefore it is highly likely that the rare variants described here may have gone unidentified were it not for a gene panel testing strategy [1]. Patients such as those described in this publication are particularly good candidates for this testing approach, since peripheral neuropathies are a clinically and genetically heterogeneous group of disorders. Using NGS technology to simultaneously analyse a large number of disease-causing genes is also more cost effective and leads to a speedier diagnosis. Obtaining a genetic diagnosis is not only beneficial to the patient in receiving a definite clinical classification and guiding appropriate treatment, but it is also important to the wider family in allowing for accurate genetic risk assessment. While not always possible, segregation analysis by testing the relatives of a patient is also invaluable to assist with interpreting the pathogenicity and clinical significance of novel variants.
In this study we identified 4 novel GARS variants previously unreported in the literature. These are likely to be associated with the disorders presenting in these patients, however further functional or segregation studies would help to verify these findings. Detailed clinical and neurophysiological information is crucial in enabling candidate genes to be identified and variants to be accurately interpreted. There are currently limitations when identifying variants that have not been studied before, however as informatics tools improve and databases expand this task is likely to become more accurate and consistent.
Identification of novel variants such as those described here expands the current knowledge of the GARS gene. Along with observations regarding the age of onset, the additional clinical features described for some patients in this study also highlights the fact that GARS variants can be associated with broad clinical phenotypes and varying degrees of severity.
Footnotes
ACKNOWLEDGMENTS
RH is a Wellcome Trust Investigator (109915/Z/15/Z), who receives support from the Wellcome Centre for Mitochondrial Research (203105/Z/16/Z), Medical Research Council (UK) (MR/N025431/1), the European Research Council (309548), the Wellcome Trust Pathfinder Scheme (201064/Z/16/Z), the Newton Fund (UK/Turkey, MR/N027302/1).