
Abstract
Background:
Hereditary muscle disorders are clinically and genetically heterogeneous. Limited information is available on their genetic makeup and their prevalence in India.
Objective:
To study the genetic basis of prevalent hereditary myopathies.
Material and methods:
This is a retrospective study conducted at a tertiary care center. The study was approved by the institutional ethics board. The point of the collection was the genetic database. The genetic data of myopathy patients for the period of two and half years (2019 to mid-2021) was evaluated. Those with genetic diagnoses of DMD, FSHD, myotonic dystrophies, mitochondriopathies, and acquired myopathies were excluded. The main outcome measures were diagnostic yield and the subtype prevalence with their gene variant spectrum.
Results:
The definitive diagnostic yield of the study was 39% (cases with two pathogenic variants in the disease-causing gene). The major contributing genes were GNE (15%), DYSF (13%), and CAPN3 (7%). Founder genes were documented in Calpainopathy and GNE myopathy. The uncommon myopathies identified were Laminopathy (0.9%), desminopathy (0.9%), and GMPPB-related myopathy (1.9%). Interestingly, a small number of patients showed pathogenic variants in more than one myopathy gene, the multigenic myopathies.
Conclusion:
This cohort study gives hospital-based information on the prevalent genotypes of myopathies (GNE, Dysferlinopathy, and calpainopathy), founder mutations, and also newly documents the curious occurrence of multigenicity in a small number of myopathies.
Keywords
INTRODUCTION
Hereditary muscle diseases epitomize the clinical and genetic heterogeneity of neuromuscular disorders. With the advent of genetic testing, these disorders are being increasingly documented. Studies describing the prevalent genotypes of myopathies in various populations are available [1–4], but the data from the Indian population is sparse; partly because genetic investigations have become available to India only recently. The vast diversity of the Indian population with its diverse ethnicity and cultural factors markedly influences the genotypic variation of myopathies. The customary practices of consanguineous marriages in parts of our population propagate these disorders through the generations. It is important to know about the prevalence of various hereditary myopathies, which paves the way to the efforts towards counseling, prevention, and development of novel therapeutic strategies.
In this study, we attempt to develop insights into these complex genetic myopathies seen in the studied population, which can further guide us to diagnose and treat them better.
MATERIAL AND METHODS
Study design
This is a retrospective evaluation of patients seen at a tertiary care center over two and a half years (January 2019 to June-2021). The study was approved by the institutional ethics board.
Inclusion criteria
Genetically tested consecutive index cases of myopathy patients were documented.
Exclusion criteria
Patients with a confirmed diagnosis of Duchenne muscular dystrophy (DMD), facioscapulohumeral muscular dystrophy (FSHD), myotonic dystrophy (DM1 and DM2), mitochondrial myopathy, and acquired myopathies
Clinical evaluation
The clinical details including the age of onset, the pattern of weakness (proximal or distal or both, upper limb or lower limb or both), and the progression was noted. From the clinical details derived, patients were categorized as those with specific and unspecified phenotypes. Serum creatine kinase levels and findings on the nerve conduction and electromyography tests were recorded.
Molecular diagnosis by exome sequencing
Exome sequencing was performed in a Clinical Laboratory Improvement Amendments and College of American Pathologists (CLIA-CAP)-certified laboratory. Peripheral intravenous blood was collected from all the enrolled patients into 10-ml commercial tubes containing ethylene-diamine tetra-acetic acid (EDTA). Focused Exome Sequencing was performed on genomic DNA with Agilent targeted sequence capture method to enrich the clinically relevant portions of the exome. Direct sequencing of the amplified captured region is performed using 2*100 bp reads on Illumina Next-generation sequencing (NGS) systems at a mean coverage of 100x in the target region. Alignment to the human reference genome (hg19) is performed and annotated variants are identified in the targeted region. The analyzed region includes coding exons and 10 bp of flanking intronic region on both sides of each exon. Variants are evaluated by their reported frequency in databases such as the Genome Aggregation Database (gnomAD), Human Gene Mutation Database (HGMD), and Clinvar. Molecular diagnosis was performed by classifying variants with clinical data correlation to identify the most likely causal disease gene as per American College of Medical Genetics and Genomics (ACMG) guidelines. Single nucleotide variants (SNVs) were confirmed by Sanger sequence analysis. CNV (Copy number variation) and AOH (absence of Heterozygosity) analysis was assessed using Biodiscovery NxClinical software or the Illumina DRAGEN Bio-IT Platform, as needed. Limitations of the test are the inability to detect single and multiple exome deletions or duplications and the variants in the regions of the genome not covered such as deep intronic, promoter, or enhancer regions and areas containing a large number of tandem repeats.
RESULTS
Molecular diagnosis and demographics
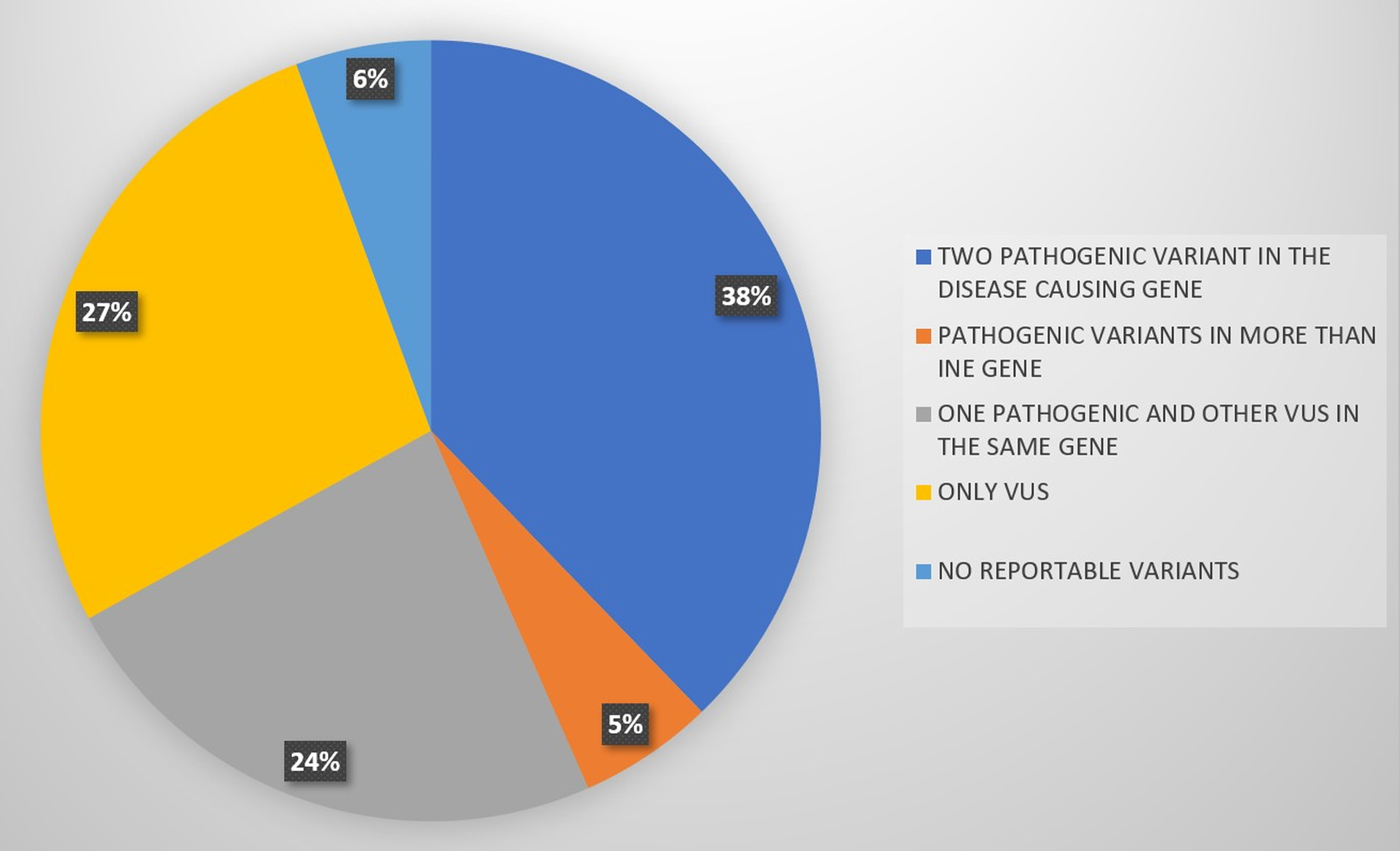
A total of 105 patients fulfilled the inclusion criteria. The mean age of the patients was 34 years, 40% were females (42/105), and 60% were males (63/105). The mean age at the time of diagnosis for the prevalent subtypes mainly GNE, DYSF, and CAPN3 were 38, 33, and 37 years, respectively. The patients were grouped as having two pathogenic mutations in disease-causing genes (N = 41) (Tables 1 and 2), one pathogenic mutation and a second VUS (N = 24), those with pathogenic mutations in more than one gene (N = 6), and those who showed only VUS in the tested genes (N = 29). The confirmed molecular diagnosis was thus established in ∼39% (41 out of 105 suspected myopathy cases), which included the confirmed cases with two pathogenic mutations in the disease-causing gene. Out of these, 4% were dominantly inherited and the rest were autosomal recessive.
Common Genotypes and its pathogenic variants found
Abbreviations: GNE UDP-N-Acetyl Glucosamine-epimerase/ N-Acetyl mannosamine kinase; DYSF Dysferlin; CAPN3 Calpain3.
Uncommon Genotypes
Abbreviations: LMNA lamin; GMPPB GDP-mannose pyrophoshphorylase B, DES desmin.
Results of NGS.
The cases (N = 24) with one pathogenic variant and the second VUS, and those with both VUS (N = 29) await supplementary genetic and molecular evaluation for further clarity. For Six patients in the cohort, no reportable variant was found on genetic testing, which is the true negative result of this study.
Major contributing genes and their allelic heterogeneity (Figs. 2 and 3)
Prevalent genotypes.
Allelic heterogeneity in the common genotypes.
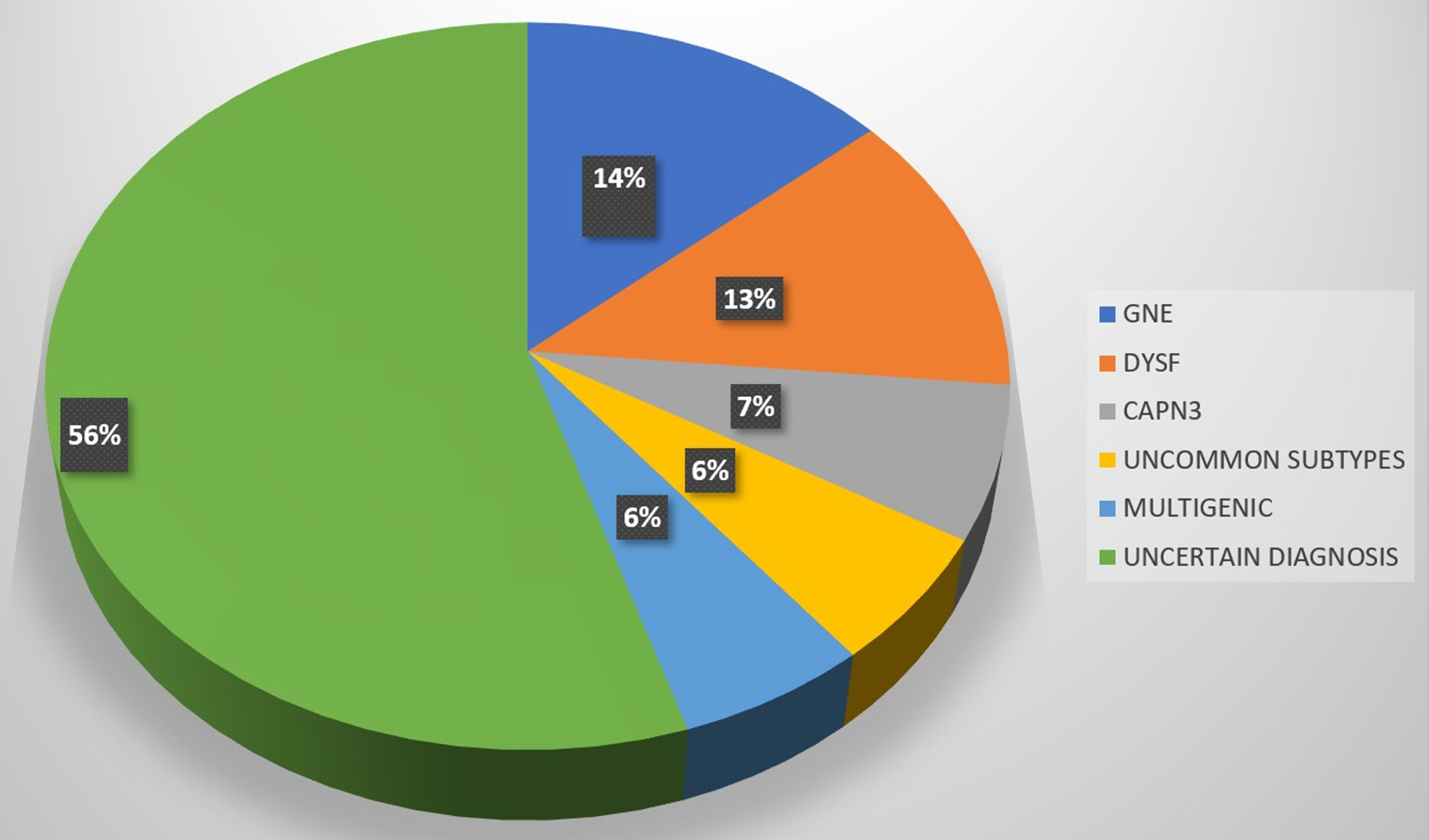
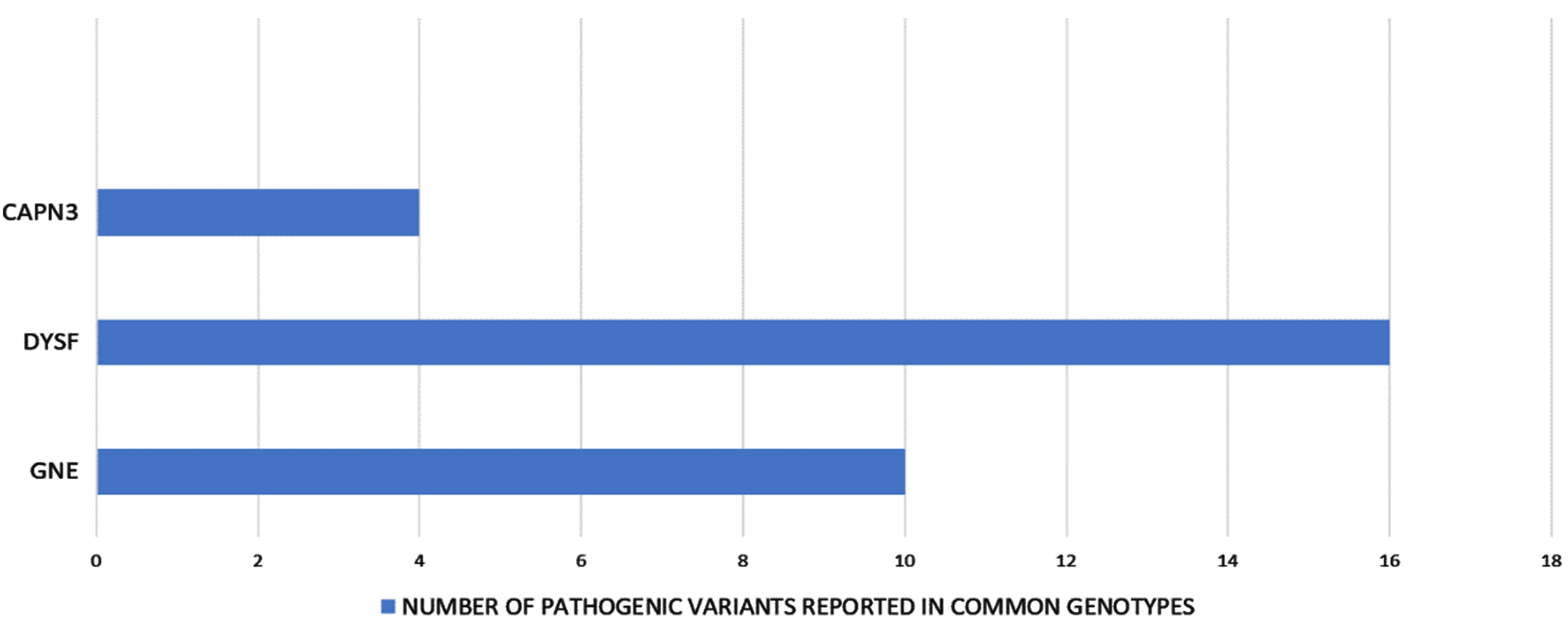
The most prevalent subtypes in this study were GNE (15%, 16/105) followed by DYSF (13%,14/105) and CAPN3 (7%,7/105). A total of 30 unique pathogenic variants were identified in all three major genes. For the GNE genotype, a total of 10 unique pathogenic variants were detected throughout the GNE gene with the majority of them being missense variants. The most frequent pathogenic variant found was c.2179G > A (p/Val727Met) and it showed the infrequent occurrence of homozygosity. The next common mutation was c.1853T > C (p.Ile618Thr) and only two deletion variants and one duplication variant was found. The next major contributing genes were DYSF and CAPN3. The DYSF genotypes were the most variegated amongst the common ones, comprising 16 different pathogenic variants staying in line with the significant allelic heterogeneity observed in this gene. The most recurring allelic variation was c.6124C > T (p.Arg2042Cys) at the exon 54. We also found one intronic variant c.5668-824C > T (at IVS50), which has been reported earlier from the Indian population. Another salient finding was a significant amount of homozygosity observed (85%, 12/14) in the DYSF genotype. Calpainopathy (CAPN3, LGMDR1) represents the most frequent LGMD-subtype worldwide but was the third most common myopathy found in the present cohort. Four unique pathogenic variants were identified and homozygosity was observed in 3 out 7 (43%) CAPN3 patients. The well-documented founder mutation in the Indian Agarwal community c.2338G > C (p.Asp780His) was present in 6 out of 7 calpain patients with 2 of them showing homozygosity.
Uncommon subtypes
These accounted for about 6% of the myopathies in this cohort. Two patients with GMPPB proteinopathy (GMPPB gene∼1.8%) and one each with autosomal dominant Laminopathy (LMNA gene, 0.9%) and Desminopathy (DES gene, 0.9%) were present.
Novel pathogenic variants
From the results of the NGS study, a total of nine novel pathogenic variants were identified in our patients (Table 3). To the best of our knowledge, these pathogenic variants have not been reported in individuals with the disease nor the general population, and are of a type expected to cause the disease, thus adding to the current literature.
Novel pathogenic variants
Clinico-genetic correlation
The phenotype-genotype correlation could be established in nearly all the patients with definitive genotypes. Patients with GNE myopathy presented with predominantly distal, anterior tibial weakness leading to foot drop, and had developed typical quadriceps sparing proximal weakness of lower limbs at the time of the examination. Another observation in our cohort was either precipitation or exacerbation of muscle weakness after childbirth in female patients [5]. Amongst DYSF genotypes, proximodistal weakness was seen most often, in various combinations of proximal and distal involvement. Calpainopathy patients in the present cohort were detected to have pelvifemoral presentation with scapular winging. Upper girdle presentations were infrequent. The majority of calpainopathy patients had tendoachilis contractures. Amongst the uncommon subtypes, two patients had cardiac involvement. The one with Laminopathy presented with limb-girdle weakness along with elbow contractures and the cardiac involvement was in form of arrhythmia. The desminopathy patient also had cardiac arrhythmias along with proximal lower limb weakness. The two other patients with GMPPB protein-related myopathy had limb-girdle weakness without any specific pattern. For the genetically uncertain cases i.e. one pathogenic and other VUS in GNE, DYSF, and CAPN3 genes, phenotypic correlated in 18 out of 24 (∼75%) cases. For example, genotypes of a single pathogenic variant in the GNE or CAPN3 gene were also associated with typical phenotypes of quadriceps sparing weakness with foot drop and scapular winging with ankle contractures, respectively. For the remainder of the genotypes in this group (LAMA2, SGCB, SCGA, ANO5, TFG), the phenotypic correlation was not very clear. For example, two patients in our cohort were shown to have a single pathogenic variant in the LAMA2 gene. The phenotype was proximal limb-girdle weakness with mild contractures of the lumbar spine and knee flexors along with myalgia and raised CK levels. One such patient had a history of consanguinity and a similarly affected sibling. Another patient with LAMA2 mutation had contractures of interphalangeal joints of the fingers. Similarly, for two other patients having single pathogenic variants of sarcoglyacanopathy genes, phenotypes were of non-specific limb-girdle weakness.
Digenic/multigenic combinations
We documented about 6% of patients as having pathogenic variants in more than one gene. Their phenotypes could not be explained by any single gene. The multigenic combinations detected were as follows: pathogenic variants each in 1) GNE and DYSF 2) GNE, SYNE1, and RYR1 3) GNE and STIM1 4) GMPPB and RYR1.
DISCUSSION
This cohort study of Indian patients provides interesting and useful genetic information. This study included a variety of inherited myopathies in patients of diverse ethnic backgrounds. As the dataset is a hospital-based evaluation, there are limitations to interpretation, but it helps give a broad idea of the genotypes seen in this region. The 39% of the diagnostic yield in our study is noteworthy and is comparable to other similar evaluations [4, 6]. The high proportion of autosomal recessive inheritance could be a reflection of the sociocultural factors and marriage customs in India.
The major contributing genes in this cohort are GNE, DYSF, and CAPN3. GNE myopathy is an autosomal recessive distal myopathy that typically shows quadriceps sparing patterns and the presence of rimmed vacuoles [7].
Initially identified in Jewish and Japanese populations [8, 9], it is being increasingly reported from all over the world with a prevalence of 1–9 per 1,000,000 [9]. In this cohort, it was the most prevalent myopathy (∼23%). To date, over 150 different GNE sequence variants have been identified in the GNE gene [9]. In the present cohort, the variant c.2179G > A (p.Val727Met) was most prevalent and is also reported as the most common and largely restricted variant to the Indian subcontinent. It is seen at a frequency of 65% in Indian GNE myopathy patients [10]. While the results in the present cohort are in agreement with the previous studies from India [10, 11], the implications of the genotype are complicated by the relatively high frequency of p.Val727Met in the normal Indian population (0.8 to 2%) [10].
From this cohort data and that of other studies [10, 11], it appears that the majority of Indian patients carry the p.Val727Met allele along with another pathogenic allele that varies from patient to patient. The frequency of occurrence of this variant underscores the scope of carrier screening for this variant in the predisposed communities, which will have an impact on the counseling and preventive aspects. The other common variant in the study c.1853T > C (p.lle18Thr) has been shown to have a strong founder effect in the Roma/Gypsy population of Europe, historically believed to be a young isolate of Asian ancestry and share this founder mutation with the north-west regions of India, particularly the Rajasthan [12].
Dysferlinopathies have a wide spectrum of phenotypes [13] and are known to account for 20% –30% of total LGMDs and around 75% of distal myopathies, especially in Asian countries [14]. Dysferlin is a large-sized gene with more than 300–400 allelic variants are reported to date [15]. Pathogenic dysferlin gene mutations were established in 16% of patients in the present cohort. Though we could identify a few recurring variants at exon 54 and 29, overall, the variants were widely spread across the entire coding sequence with no apparent mutational hot spot identified. There are seven recorded founder mutations in various populations worldwide. These have been detected in Portuguese (c.1180_1180 + 7delAGTGCGTG, c.5492G > A) [16], native Canadian (c.2745C > G) [17], Caucasian Jewish (c.2779delG) [18], Italian (c.2875C > T) [19], Lebanese Jewish (c.4872delG.fsX9) [20] and Spanish (c.6086C > T) [21] populations. But to the best of our knowledge, no such founder mutation has been reported from the Indian subcontinent to date. Further such studies from different geographical regions and ethnic backgrounds probably should provide more information [22]. However, the extensive homozygosity (85% in the present cohort) observed in this cohort dictates the immediate need for carrier testing for these subtypes.
Calpainopathy (LGMD2A) is perhaps the most common subtype of LGMD worldwide and more than 100 pathogenic mutations in the CAPN3 gene (encoding non-structural protein calpain) have been identified to date [23]. Various founder mutations have been described for small populations of Brazil, Spain, and Japan [24, 25] and in Agarwals of India as well [26]. In a recent study, a systematic characterization of the genetic framework of LGMDR1 has been done for North Indian population [27]. Our study stands somewhat differently from the above-mentioned one, in calpainopathy being less frequent than the other two major genotypes (GNE and DYSF) and showing the founder effect in our cohort, probably reflecting the regional differences within the country. Nearly all of our calpainopathy patients belonged to the Agarwal community and most of them harbored the well-described founder variant c.2338G > C (p.Asp780His) at one or two alleles. The two ancestral founder mutations in the calpain gene, a missense (c.2338G > C; p.D780H) and a splice-site (c.2099-1G > T) mutation are prevalent due to the practice of intra-communal exogamy in the community of Agarwals [26]. These founder mutations have also been reported in a similar study from South India [28]. This emphasizes the utility of targeted mutational analysis and carrier testing in select groups in resource-limited countries like India [29].
Amongst the uncommon subtypes in this cohort, there were two patients with GMPPB-related myopathy and two patients with autosomal dominant LGMD, Laminopathy, and Desminopathy. Considering the low prevalence of these uncommon subtypes, we need much larger studies to draw useful conclusions. About Sarcoglyacanopathies, our findings are not along the line of previous reports [30]. This could partially be attributable to selection bias in the cohort, the majority being the adult population. The NGS studies detected nine novel pathogenic variants which are important for the evolving database (Table 3).
Genotype-phenotype relationship
Genetic diagnosis could be predicted in the majority of the cases of GNE, dysferlinopathy, and calpainopathy, reflecting the rather homogenous phenotypes of these conditions (Fig. 4) as described in the results.
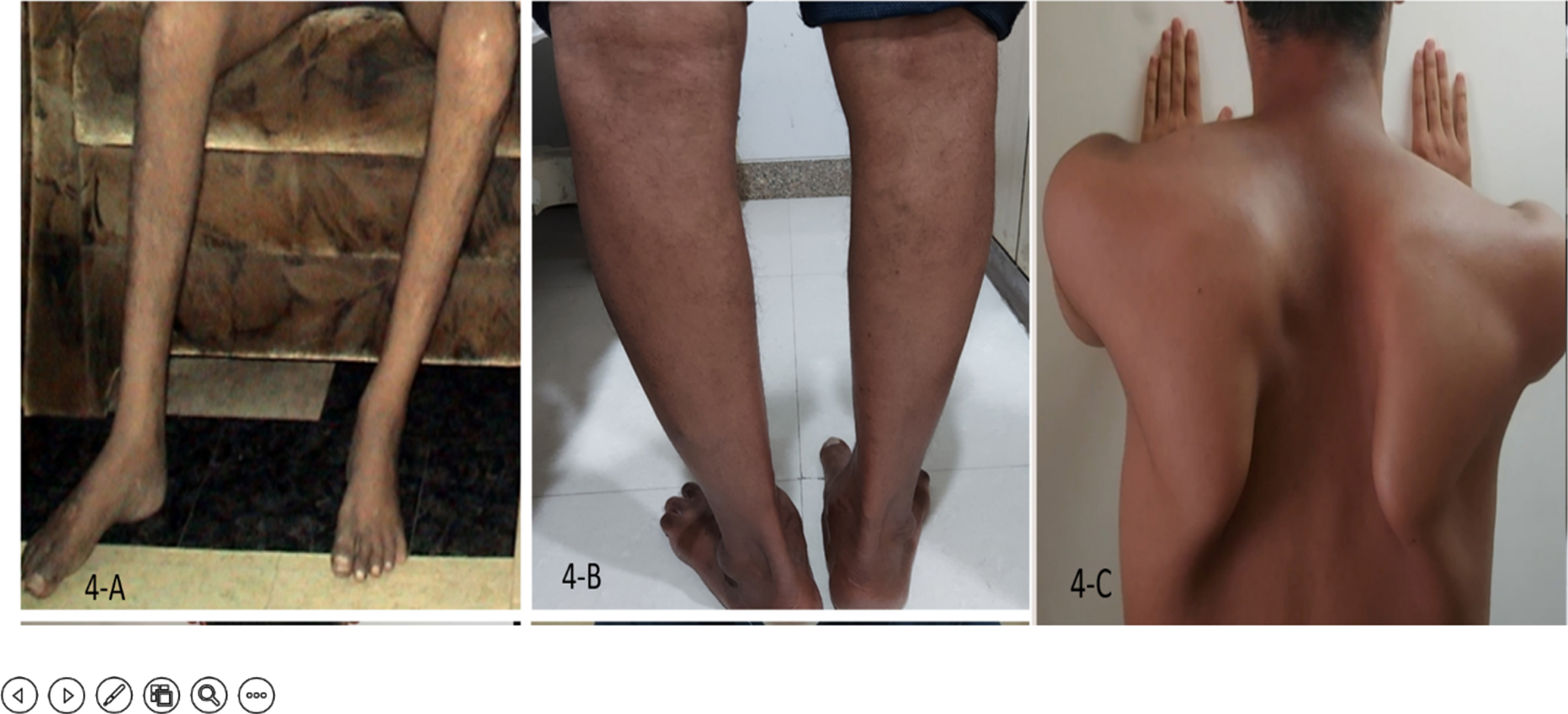
Phenotypic features. A)Bilateral foot drop and wasted tibialis anterior in GNE myopathy; B) Wasted calves in Dysferlinopathy C) Scapular winging in Calpainopathy.
There have been attempts at establishing the genotype-phenotype relationships earlier; in the GNE gene, i.e. p.Ala662Val may be associated with a more severe phenotype, compared to p.Val727Met [31]. In this cohort, while Val 727 met was outstanding in its frequency, natural history studies were not undertaken. In dysferlinopathy, the possibility of a particular pathogenic variant predicting the particular phenotype has been studied, e.g. patients with p.Tyr999Cys had LGMD phenotype much more than Miyoshi myopathy. However, no significant phenotypic distinction could be made between various mutations.
The multigenic myopathies-a new frontier
An intriguing observation from this study was a small number of patients having pathogenic alleles in more than one gene and showing unusual clinical presentations. There is very little literature available on the subject and these findings are of interest. Explanations for this occurrence could be the role of synergistic heterozygosity and digenic contribution, Epistasis meaning modulating effect of one gene over another, or mere semblance as carriers for one of the genes involved. Multigenicity as a novel disease mechanism in inherited myopathies has been reported recently in the literature [32, 33]. We document these patients in this cohort. For further understanding of these occurrences, functional platforms like RNA sequencing and proteomic studies are required to elucidate the variable expression of each of the genes at the effector organ.
CONCLUSIONS
This large cohort study documents the 39% yield of molecular diagnosis by NGS. GNE, DYSF, and CAPN 3 genotypes formed the main three groups of genetic diagnosis, and the correlation with phenotype was high. Founder mutations were encountered in GNE myopathy and Calpainopathy while none was present in dysferlinopathies. Few of the uncommon subtypes were also documented. There were patients with a single pathogenic allele detection, which requires further workup in the future. The unusual occurrence of multigenicity is reported in this cohort, again an area of potential research. With the novel therapies approaching, molecular diagnosis of hereditary neuromuscular disorders in different populations will have useful implications in terms of therapeutics, prevention as well as an understanding of the molecular mechanisms.
Footnotes
ACKNOWLEDGMENTS
None
CONFLICTS OF INTEREST
None