
Abstract
Introduction:
Titinopathies are heterogenous group of disorders affecting the skeletal and cardiac muscles variably and caused by Titin (TTN) gene mutations located in Chromosome 2. The manifestations extend from congenital to adult-onset myopathies. Here we describe the phenotype-genotype heterogeneity of patients with myopathy/muscular dystrophy associated with TTN variants in an Indian cohort.
Methods:
A retrospective descriptive study of 12 patients diagnosed with primary muscle disease evaluated between 2016 and 2023 harboring rare TTN variants.
Results:
Eight patients were included (M:F ratio - 3:1). The median age at onset of entire cohort is 5 (range: birth- 33 years). The major clinical phenotypes were congenital myopathy [n = 3, 37.5%], juvenile onset myopathy [n = 3, 37.5%] and adult AD - Hereditary myopathy with early respiratory failure (HMERF) phenotype [n = 2, P6, P8; 25%]. Prominent / wide first interdigital space in feet in congenital and juvenile forms (c.38421_38437delinsC, c.106531 + 1G > A) was a novel feature. The variant c.95134T > C previously reported in HMERF in British population, was noted in two patients in our cohort and with GNE myopathy like phenotype in one. Muscle MRI done in congenital myopathy (c.26201-1G > A) showed fatty infiltration of anterior and posterior thigh with sparing of gracilis, adductor magnus and tibialis anterior.
Conclusion:
This is the first Indian study with a large cohort demonstrating many novel mutations and clinical heterogeneity expanding the spectrum of titinopathies.
Introduction
Titin is the largest protein and key component of contractile apparatus of the muscle fibre. It extends from the Z-disc to the M-Band of the sarcomere along with myosin and actin. 1 Titin (TTN), the largest gene residing in chromosome-2 has a coding sequence of more than 100 kb of cDNA. Mutations in TTN cause heterogenous skeletal muscle and cardiac diseases. Most of these variants are reported to be associated with isolated cardiomyopathy than skeletal myopathy. 2 The various types of titinopathies extend from congenital to adult onset, autosomal recessive (AR) to autosomal dominant (AD) forms. 3 The congenital forms are recessive titinopathies with or without cardiomyopathy, respiratory insufficiency and developmental delay with contractures. Juvenile onset types are Limb girdle muscular dystrophy (LGMD-R10), Emery-Dreifuss like myopathy without cardiac involvement and early onset distal myopathy. Adult onset titinopathies are predominantly dominant phenotypes including Tibial muscular dystrophy (TMD) and Hereditary myopathy with early respiratory failure (HMERF) while recessive distal or proximal myopathies have been rarely reported. 4 Following the advent of next generation sequencing, there has been an expansion of various phenotypic manifestations with various novel mutations. Previously, only three cases have been reported from India.5,6 In this study we describe the phenotype-genotype heterogeneity of patients with myopathy/muscular dystrophy associated with TTN variants in an Indian cohort.
Methods and materials
This is a retrospective descriptive study conducted at a neuromuscular division of a quaternary hospital for neurological disorders in India on patients with clinical features of myopathy and genetic variants identified in TTN gene. The baseline demographic details, laboratory findings including muscle MRI, muscle histopathology details were collected from the medical records and analyzed. Fatty infiltration of muscles was described using modified Mercuri grading. 7 Muscle histopathological details included routine eosin and hematoxylin, modified Gomori's and oxidative stains. All patients underwent next generation sequencing as previously described. 8 TTN variants identified were classified as per ACMG guidelines using variant interpretation tool Franklin by Genoox (https://franklin.genoox.com). 9 The transcript ID for TTN gene used is NM_001267550.2 and exon numbering was done based on LRG_391 numbering recommended for clinical reporting (https://www.cardiodb.org/titin/titin_transcripts.php). All data were analyzed and presented as descriptive statistics. Informed consent was obtained from all the patients and ethical approval was obtained from the Institution ethics committee [IEC no: NIMH/DO/(BS&NS) 2022]. Descriptive statistics such as mean, median and range were used to describe the data.
Results
Demography: Eight patients with TTN pathogenic variations were included in this study. The median ages at onset of symptoms and presentation were 5 (range: birth- 33 years) and 16.5 (range: 7months – 34 years) respectively. The M:F ratio is 3:1.
Clinical features
Shown in Table 1.
Clinical characteristics of the patients with titinopathies.

The most common presenting symptom was proximal lower limb (LL) weakness (n = 5) followed by delayed motor milestones (n = 3). One patient also had additional distal LL weakness (P8) at presentation. Patients P1 and P3 had history of reduced fetal movements along with respiratory distress at birth. Two patients had distal upper limb (UL) weakness (P5, P6). None of the patients or their family members had symptoms suggestive of cardiac involvement. History of consanguinity was noted in 5 patients. However, none had significant positive family history. The most common muscle involved includes neck flexors, gluteus maximus, hamstrings and tibialis anterior. One patient had selective thenar muscle weakness and wasting (P5) (Figure 1a, b). Pattern of muscle involvement is depicted in Supplementary figure 1. Two patients required assistance for ambulation at presentation (P2, P6). The median creatine kinase (CK) level was 1261 IU (range: 95–2984). All patients underwent electrocardiogram and echocardiography at initial assessment which was normal. Muscle MRI was available in 1 patient (Figure 2). All underwent clinical exome sequencing and results are elaborated in Table 2.
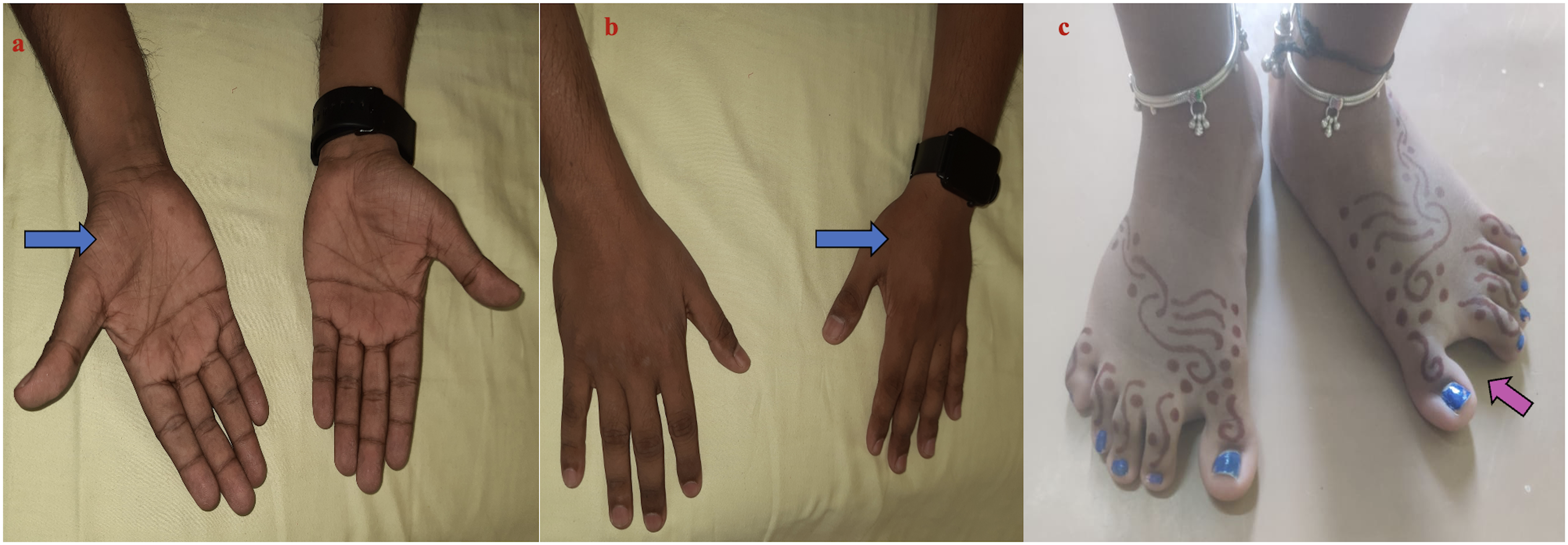
Clinical images of patients: a: Thenar (blue arrow) and b: first dorsal interossei wasting (P5) (blue arrow). c: Increased first interdigital space of the feet (P1).
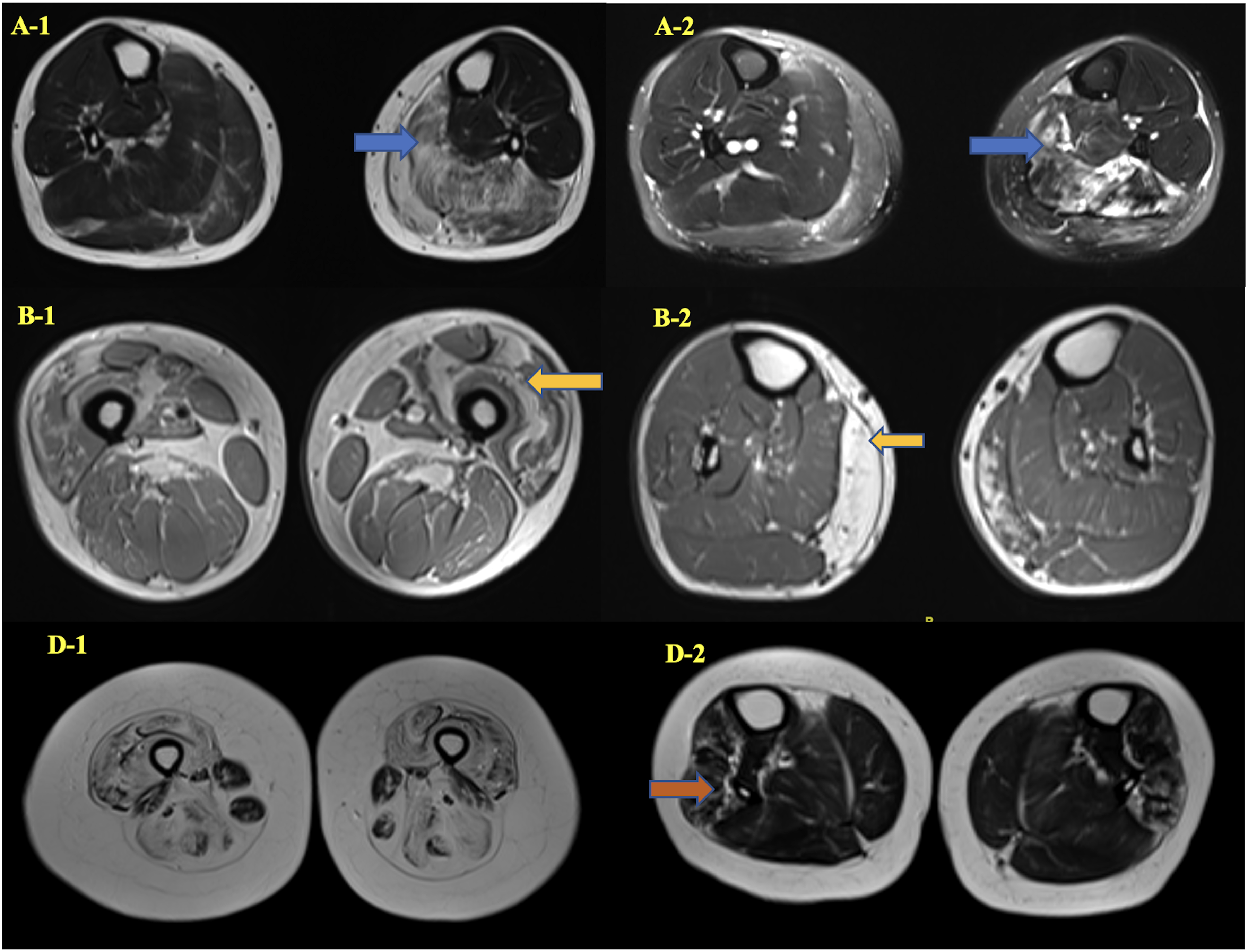
Muscle MRI of patients: gastrocnemius and soleus (L > R) (blue arrow) showing fatty infiltration in T1W: A-1 and STIR hyperintensity (blue arrow): A-2 (P9). Quadriceps (L > R) (yellow arrow) fatty infiltration in B-1 and medial gastrocnemius (R > L) (yellow arrow) in B-2 (P10). Diffuse fatty infiltration of anterior and posterior thigh with sparing of gracilis and adductor magnus in D-1, tibialis anterior in D2 (P2).
Genotype – phenotype characteristics.

Clinical phenotypes: The major clinical phenotypes were congenital myopathy [n = 3: P1, P2, P3; 37.5%], juvenile onset myopathy [n = 3, P4, P5, P7; 37.5%] and adult AD - HMERF phenotype [n = 2, P6, P8; 25%].
Spectrum and location of genetic variants observed in the study are shown in Figure 3.
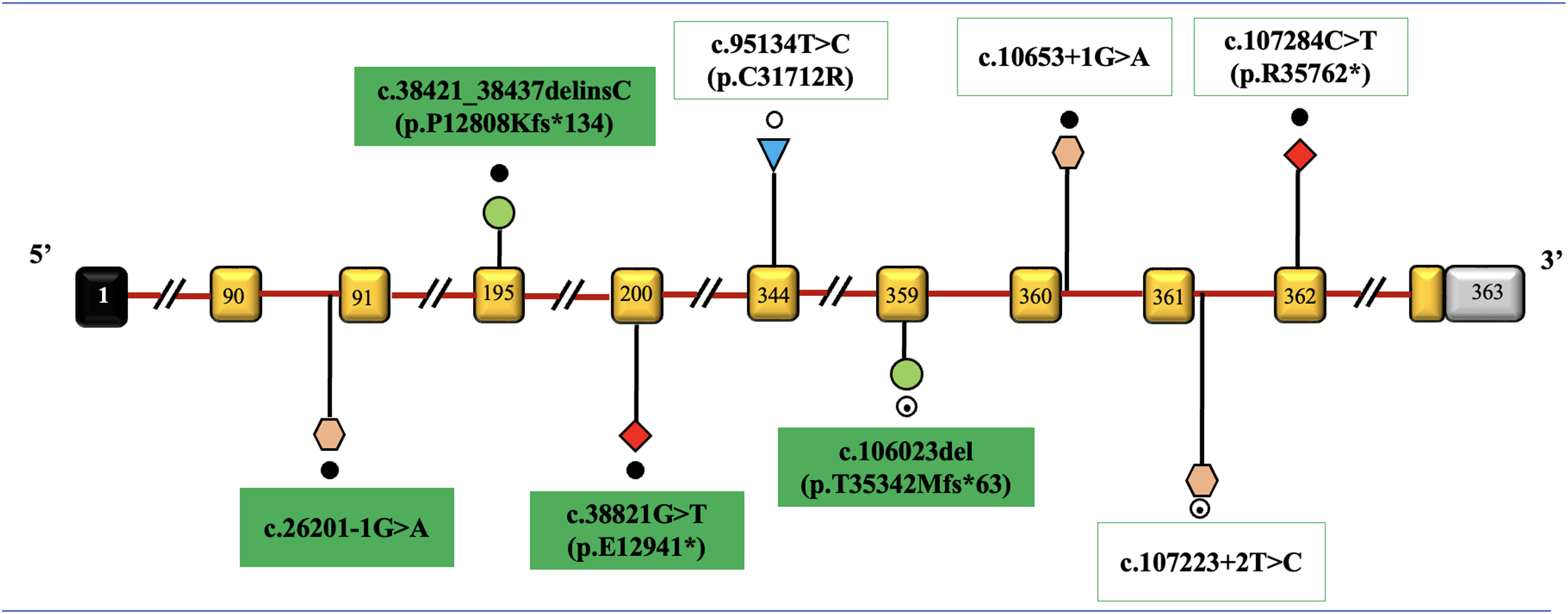
Spectrum and location of genetic variants observed in the study: TTN gene (transcript ID: NM_001267550.2). The exons are represented as rectangular boxes with curved edges with respective exonic numbers with non-coding regions shaded in black and grey at the ends. The novel variants are shaded with green. The dark dots represent homozygous variations, unfilled dots represent heterozygous variations, semi filled dot represent compound heterozygous variations. Triangles represent missense, rhombus represent nonsense, circles represent frameshift, and hexagon represent intronic variations. Size of exons /introns is not represented at scale.
Discussion
Titinopathies are a heterogenous group of muscle disorders which may be associated with cardiomyopathy. This is the largest series of titinopathies being described from India.
The clinical features of congenital titinopathy were comparable to the study done by Oates et al., on a large cohort of 30 patients. 11 However, the presence of velvety soft palms and soles noted in two of our patients is not reported previously. All the patients in the juvenile onset group had severe proximal weakness with multiple joint contractures. Similar phenotype of LG syndrome with prominent joint contractures without cardiac involvement has been reported by earlier authors.12,13,14 Increased 1stinterdigital space of feet noted in congenital / juvenile forms was a novel clinical feature.
MRI muscle features of adult-onset patients had predominant fatty infiltration of posterior legs involving the medial gastrocnemius, soleus and flexor hallucis longus and this is also a novel finding. The common muscles involved includes gluteus minimus, sartorius, semitendinosus and tibialis posterior.3,15 Study by Evila et al., showed that adult-onset patients showed fatty infiltration of anterolateral legs and soleus along with hamstrings and glutei muscles. 16 In the congenital onset group muscle MRI features was comparable to the study by Oates et al., 11 with quadriceps and hamstrings being affected, however, involvement of tibialis anterior identified in our study is not reported earlier.
In our patients with confirmed disease-causing TTN mutations, most presented with congenital myopathy (3) or juvenile onset myopathy (3) and associated with recessive genotypes. Two patients had adult-onset myopathy with dominant genotype. The total number of homozygous variants in the current cohort is 5 with history of consanguinity noted in 5 patients. While truncating variants in proximal I or A bands have been predicted to cause Nonsense Mediated Decay (NMD) with loss of full protein resulting in severe recessive phenotypes, distal truncations in M-band (exons 359–363) might escape NMD with residual near full length protein expression. 12 Recessive truncating mutations especially in C-terminal region especially in the terminal two exons have been associated with milder proximal / distal phenotypes with later onset.12,17 In our cohort all truncating variants except in P4 were present in proximal exons and associated with congenital phenotype, while P4 with homozygous nonsense variant c.107284C > T (p. Arg35762Ter) affecting exon 362 presented with adult-onset phenotype. Canonical splice site variants have been identified in three patients and interestingly in two patients (P5 and P7) with juvenile onset myopathy, the variants affected donor splice sites of distal exons of 359 and 360 respectively in crucial M-band. In juvenile myopathy group in contrast to previous studies, two patients had involvement of 5’ splice site mutation which is not reported earlier.17,18
In P2 a splice acceptor variant affecting proximal I-band exon 91 resulted in a more severe congenital onset phenotype. Of the 5 truncating variants all are novel except in P4, which is previously reported in TTN related dilated cardiomyopathy. 18 There was no evidence of cardiac involvement in all patients from the three groups. A study by Savarese et al., 12 described 23 AR titinopathy cases with 10 congenital and 13 non-congenital phenotypes. Most common involvement was noted in M-band followed by I-band. Congenital forms had common involvement of meta-transcript only pathogenic variant and splice site involvement. All patients with non-congenital forms had involvement of exons 363, 364 and intron 362. Non-congenital myopathies had involvement of exons near c-terminal and congenital forms near N-terminal regions.
The most common form of adult-onset AD myopathy reported is Tibial Muscular Dystrophy (TMD) followed by Hereditary myopathy with early respiratory failure (HMERF). 3 The two adults reported by Nalini et al., had limb girdle weakness along with severe distal anterior leg weakness and wasting. 5 Interestingly, two patients (P6, P8) with adult-onset AD myopathy in our cohort had the HMERF variant c.95134T > C (p. Cys31712Arg) in the hotspot exon 344 which is most commonly reported in British population. 19 Previously only one patient of Indian origin is reported with this variant. 20 Onset of symptoms is usually in the 3rd – 4th decade with proximo-distal limb and axial weakness with early respiratory failure. In the present study patient P6 had distal UL and proximal LL weakness whereas P8 had distal followed by proximal LL weakness. Though both had onset of symptoms in the 3rd decade, P8 was slow in walking and running since childhood. In addition, P8 also had GNE myopathy like phenotype with neck flexor wasting and weakness and hamstrings being weaker than quadriceps which is a novel finding. Both the patients did not have respiratory involvement.
Conclusion
The large size and complexity of TTN gene results in varied genotype and phenotype heterogeneity as also observed in the present study. Our study shows a heterogenous spectrum of characteristics with respect to age of onset, disease course and many novel variants expanding the spectrum of titinopathies.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602241313119 - Supplemental material for Titinopathies: Phenotype – genotype heterogeneity in an Indian cohort
Supplemental material, sj-docx-1-jnd-10.1177_22143602241313119 for Titinopathies: Phenotype – genotype heterogeneity in an Indian cohort by Dipti Baskar, Seena Vengalil, Kiran Polavarapu, Veeramani Preethish-Kumar, Saraswati Nashi, Gautham Arunachal, Kosha Srivastava, Vaishnavi Desai, Priya Treesa Thomas, Muddasu Suhasini Keerthipriya, Akshata Huddar, Gopikrishnan Unnikrishnan, Ram Murthy Anjanappa and Atchayaram Nalini in Journal of Neuromuscular Diseases
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.