
Abstract
Background and Purpose:
Mutations in the GMPPB gene affect glycosylation of α-dystroglycan, leading to varied clinical phenotypes. We attempted to delineate the muscle MR imaging spectrum of GMPPB-related Congenital Myasthenic syndrome (CMS) in a single-center cohort study.
Objective:
To identify the distinct patterns of muscle involvement in GMPPB gene mutations.
Methods:
We analyzed the muscle MR images of 7 genetically proven cases of GMPPB dystroglycanopathy belonging to three families and studied the potential qualitative imaging pattern to aid in clinico -radiological diagnosis in neuromuscular practice. All individuals underwent muscle MRI (T1, T2, STIR/PD Fat sat. sequences in 1.5 T machine) of the lower limbs. Qualitative assessment and scoring were done for muscle changes using Mercuri staging for fibro-fatty replacement on T1 sequence and Borsato score for myoedema on STIR sequence.
Results:
All patients were of South Indian origin and presented as slowly progressive childhood to adult-onset fatigable limb-girdle muscle weakness, elevated creatine kinase level, and positive decrement response in proximal muscles. Muscle biopsy revealed features of dystrophy. All patients demonstrated identical homozygous mutation c.1000G > A in the GMPPB gene. MRI demonstrated early and severe involvement of paraspinal muscles, gluteus minimus, and relatively less severe involvement of the short head of the biceps femoris. A distinct proximo-distal gradient of affliction was identified in the glutei, vasti, tibialis anterior and peronei. Also, a postero-anterior gradient was observed in the gracilis muscle.
Conclusion:
Hitherto unreported, the distinctive MR imaging pattern described here, coupled with relatively slowly progressive symptoms of fatigable limb-girdle weakness, would facilitate an early diagnosis of the milder form of GMPPB- dystroglycanopathy associated with homozygous GMPPB gene mutation.
INTRODUCTION
GDP-mannose pyrophosphorylase B (GMPPB) related phenotype spectrum ranges widely and inc-ludes congenital myasthenic syndrome (CMS), limb-girdle muscular dystrophy (LGMDR19), and severe disorders of α-dystroglycanopathy [1–4].
LGMDR19 is caused by homozygous or compound heterozygous mutations in the GMPPB gene, which encodes the beta subunit of GDP-mannose pyrophosphorylase on chromosome 3p21. It is an autosomal recessive muscular dystrophy characterized by early childhood onset mild proximal muscle weakness and less frequent additional features of mild intellectual disability or seizures. It is part of a group of similar disorders resulting from defective glycosylation of alpha-dystroglycan, collectively known as ‘dystroglycanopathies’ [1–3].
Carss et al., have reported on 8 cases of GMPPB related mutations with a wide spectrum of presentation ranging from classical CMD associated with ataxia, absent speech development, and brain abnormalities to children with mild limb-girdle weakness and mild intellectual disability to isolated LGMD phenotype [1]. Some patients with GMPPB mutations may show features consistent with a congenital myasthenic syndrome, with fatigability and decremental compound muscle action potential response, and these patients can demonstrate a positive therapeutic response to treatment with pyridostigmine [5, 6].
Overall, about 81 patients harbouring mutations in the GMPPB gene have been described worldwide: 56 with LGMD or overlapping LGMD-CMS phenotypes and the remaining with features of CMD [4]. The milder variants present in adulthood, with limb-girdle muscle weakness (LGMW), isolated rhabdomyolysis, or CMS features [7, 8].
Currently, muscle MRI has enabled recognition of particular patterns of involvement typical of various inherited muscle disorders.. There are only a few studies briefly mentioning the MRI pattern of muscle involvement in GMPPB-LGMD-CMS phenotype secondary to compound heterozygous mutations [3, 9].
For the first time, we have identified a distinct pattern of muscle involvement on MRI in 7 patients belonging to 3 families with an identical homozygous mutation in the GMPPB gene. This finding could assist in identifying and differentiating GMPPB-LGMD-CMS from other LGMDs’ and aiding in early diagnosis and targeted gene testing.
METHODS
All patients provided written informed consent to publish their medical data, clinical photographs, and MR images. Institute Ethics Committee approval has been obtained for the study. A prospective evaluation of clinical characteristics, biochemical, histopathological, genetic, and muscle MRI findings in 7 patients belonging to 3 families are being described. All patients were evaluated at a quaternary national institute for neurological disorders. The three families hailed from southern India. A thorough clinical examination was done by the neuromuscular specialist (AN), and all findings were recorded in a pre-designed proforma. Electrophysiological studies were performed as per the standard protocol for nerve conduction studies. Repetitive nerve stimulation (RNS) for decrement (orbicularis oculi, nasalis, trapezius, deltoid, quadriceps, and TA muscles) was performed at a 3 Hz stimulation rate. Anconeus muscle was also tested for decrement (P6). The femoral nerve was stimulated immediately below the inguinal ligament just lateral to the femoral artery pulsation. The common peroneal nerve was stimulated at the fibula head for recording from the TA muscle. Post-exercise testing was not done (supplementary data). Concentric needle electromyography from biceps brachii and quadriceps muscles was done. Quadriceps muscle biopsy was performed in three patients (Patient 1, 5, 7). After the genetic confirmation, patients were subjected to muscle MRI of the lower limbs.
Gene testing
Genetic testing was performed by muscular dystrophy panel sequencing (53 genes) in all members from family 1, while the remaining patients underwent clinical exome sequencing.
Muscle MRI protocol
Muscle MRI of lower limbs with 5 mm axial sections extending from the iliac crest up to the lateral malleoli was performed on a 1.5 Tesla machine (SIEMENS) as a multi-sequence imaging protocol and included T1-weighted (T1W) and T2-weighted (T2W) (turbo) spin-echo, as well as Fat, suppressed sequences (STIR/PD FS). The coronal images of the thorax were available in patient 2 of family 1. Two neuroradiologists (CP, SS) and a neurologist independently analyzed and graded the MR images for fat infiltration by Mercuri scoring and for myo-edema on the fat-suppressed images by Borsato scoring [10, 11]. Fatty infiltration was graded using the 5-point scale with scores ranging from 0 (normal appearance) to 4 (complete fatty replacement). In case of discordance between the observers, an agreement was reached by consultation. A cumulative score per patient (T1-MRI score) and a mean T1 score per muscle were calculated.
RESULTS
Clinical and electrophysiology data
The clinical details are summarized in Table 1. All patients shared the common features of gradually progressive limb-girdle weakness, with preferential muscle involvement and prominent truncal muscle weakness (Fig. 1-I). The majority had hyperactive tendon reflexes. There were four men and three women belonging to 3 families. Consanguinity was present in families 1 and 2, while patient 7 was born to non-consanguineous parents (Fig. 1.1). The onset of symptoms occurred from childhood to 40 years of age. The duration of illness ranged from 7 to 40 years. The illness progression was slow, and the majority were ambulant at presentation. All ind-ividuals had a progressive fatigable limb-girdle weakness with difficulty in walking fast, climbing stairs, walking uphill, rising from the floor, and buckling with falls. Although persistent limb-girdle muscle weakness was a characteristic finding, they also had fluctuating and fatigable muscle weakness with diurnal/weekly/seasonal fluctuations (warm/hot weather), which also worsened during episodes of fever and menstruation suggestive of neuromuscular junction defect. The fluctuations were spread over days to weeks in most of them. However, the muscle fatigue was not profound and usually appeared after prolonged physical exertion with the regaining of strength after rest. None had cardiac or respiratory symptoms.
Salient clinical features of the patients
On examination, all patients had calf hypertrophy, variable ankle contractures, and preferential pattern of limb-girdle weakness typically observed in limb-girdle muscular dystrophy (LGMD). Mild ptosis was present in the siblings (P5, P6) belonging to family 2. The muscle strength varied depending on the duration of the illness (modified MRC grade 2 to 4). Selective severe weakness of the pectoral and pelvic girdles, wasting of shoulder girdles, and thigh muscles with scapular winging with increasing duration of illness were noted. Distal muscles were spared except for mild weakness in the severely affected individuals. Overall the tendon reflexes ranged from being hypoactive to hyperactive.
Nerve conduction studies demonstrated a clear decrement of compound muscle action potential on slow, repetitive nerve stimulation (3 Hz) performed in the resting proximal large muscles. As patients predominantly had a limb-girdle weakness, lower limb muscles like quadriceps and TA in addition to trapezius and deltoid were selected for RNS. The femoral and common peroneal nerves were stimulated, and CMAPs were recorded. The procedure was well tolerated by the patients. Prominent decrement was demonstrated in the proximal muscles tested and ranged from 25%–67%(Fig. 1-H). No significant decrement was observed in the facial and distal muscles, which were not clinically affected. Concentric needle electromyography of Biceps, Quadriceps, and TA showed evidence of short duration, low amplitude polyphasic motor unit potentials suggestive of a myopathic process in all patients.
Biochemical findings
The creatine kinase level was elevated in all and ranged from 714 to 6220 IU / L. The lowest level was seen in the patient with the most advanced disease (P1). The liver enzymes were mildly elevated (range 56 to 74 mg/dL).
Histopathology findings
Open muscle biopsy from the quadriceps was performed in three patients. The muscle biopsy showed varying changes on histopathological examination, which ranged from neurogenic changes to dystrophic changes. Patient 1 (Fig. 2A, D, G) showed prominent myopathic changes that included early effacement of architecture, variation in fibre size, rounding of fibres, splitting, internalized nuclei, scattered hypertrophic, and atrophic fibres with interstitial fibrosis. Focal perivascular inflammation is noted, as indicated in Fig. 2D (arrow). There was a questionable presence of tubular aggregates. As the tissue was, fixed in 10%formalin, enzyme histochemistry (oxidative enzyme and ATPase stains) and immunohistochemistry could not be performed. Patient 5 (Fig. 2B, E, H, K, L) showed mild variation in fibre size with a few angulated atrophic fibres and clumped nuclei, suggesting mild neurogenic changes. ATPase stain performed on this (Figure K, ATPase 9.4; Figure L. ATPase 4.6) indicated type II fibre predominance suspicious of type II fibre grouping. Patient 7 (Fig. 2C, F, L and I) showed myopathic features, which were less distinct as compared to the first case with preserved fascicular architecture, mild variation in fibre size, and a few regenerating fibres (Fig. 2C). On modified Gomori trichrome stain, additionally variation in fibre size and mild interstitial (endomysial) fibrosis was noted. The NADH stain performed on Patient 5 (Fig. 2E) and Patient 7 (Fig. 2F) showed dark hyper-stained granular areas which were not appreciated (not stained) on SDH stains (Fig. 2 H and I). The nature of this granular material is not clear and could not be further characterized as electron microscopy was not possible. Since they were highlighted only by NADH stain, with SDH stain not demonstrating them, these granules could probably represent/suggest (but not definite/unconfirmed) tubular aggregates.
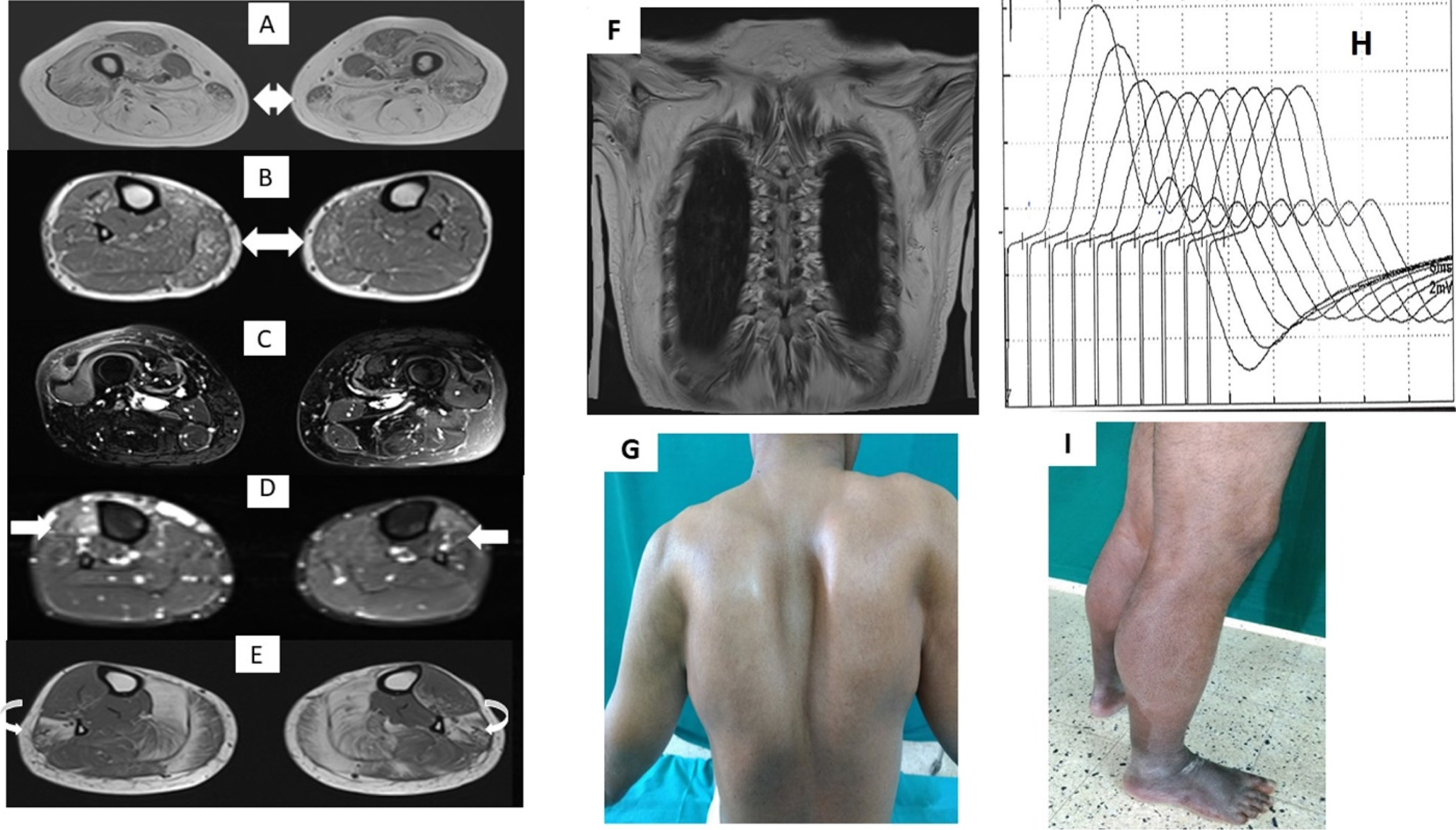
(A): Double headed arrow shows the postero-anterior gradient within gracilis in first patient (P5) of family 2. (B):Double headed arrow show less severe involvement (when compared to other families) of the medial gastrocnemius muscle in the patient of family 3.(C,D): Arrows show segmental myoedema in right quadratus in second patient (P6) of family 2 and TA edema in the patient (P7) of family 3 respectively. (E):Curved arrow shows preferential gradient in the peroneal group with severe involvement of peroneus brevis when compared to longus. (F.G):Coronal T1 MRI of first patient (P1) of family 1 depicting severe fatty infiltration of scapular muscles with a corresponding clinical photograph depicting the winging of scapula consequent to it. (H):Repetitive Nerve stimulation of femoral nerve shows prominent decrement in Quadriceps muscle. (I): Prominent calf hypertrophy is seen in second patient (P2) from family 1.
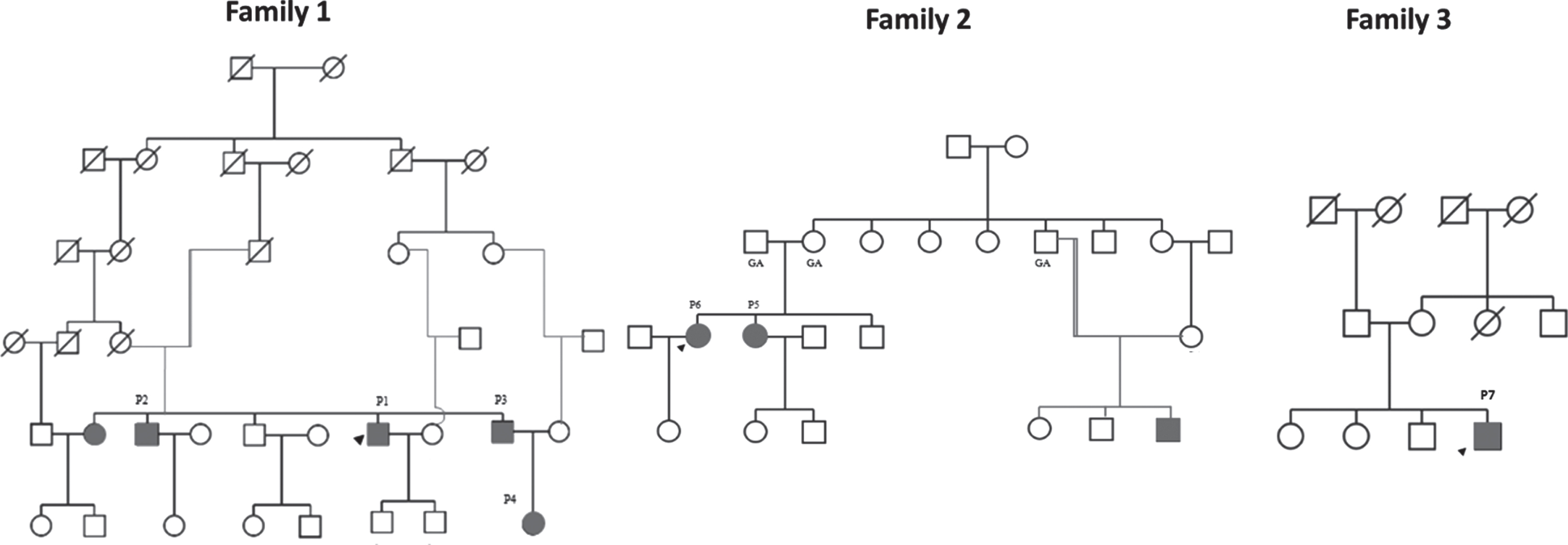
Pedigree diagrams of the families under study. The affected individuals who are included in the study are number as P1 to P7. The square symbol represents male, circle female, solid affected individuals, and unfilled symbols for unaffected individuals.
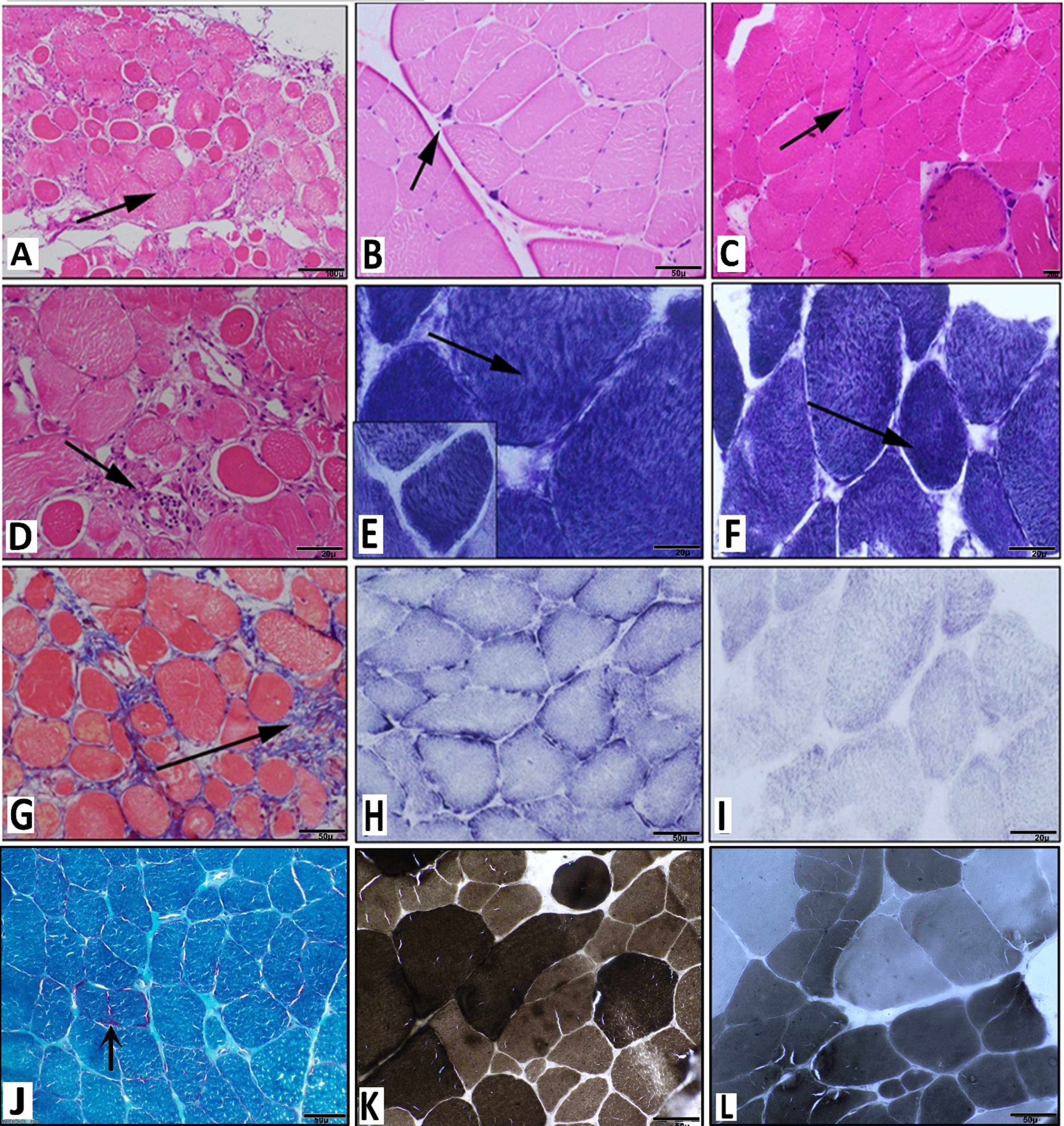
A: Microphotograph showing transversely sectioned muscle tissue with variation in fibre size, partial effacement of architecture, rounding of fibres with hypertrophy (arrow) and atrophy of fibres. [H & E stain, x100]. B: Microphotograph showing muscle tissue with reserved fascicular architecture but with neurogenic changes (hypertrophic fibres, angulated atrophic fibre and clumped nuclei – arrow). [H & E stain, x100]. C: Microphotograph showing variation in fibre size including hypertrophic fibres, angulated atrophic fibre, fibre splitting, internalised nuclei and regenerating fibres (arrow) [H & E x200]. Inset shows one of the fibre with basophilic granular stippling (H & E x400). D: Microphotograph showing transversely sectioned muscle tissue with variation in fibre size, partial effacement of architecture, rounding of fibres with hypertrophy (arrow) and atrophy of fibres. [H & E stain, x200] Also seen in this image is focal perivascular inflammation (arrow). E,F: NADH stain (Oxidative enzyme) stain (x200) showing muscle fibres showing variation in fibre size with scattered hypertrophic fibres along with dark hyperstained granules (arrow) better observed in the inset image (E, inset×400). G: Microphotograph (Masson Trichrome stain) highlighting the interstitial fibrosis (arrow) and also the variation in fibre size with rounding of fibres. H,I: SDH stain showing muscle with mild variation in fibre size and without any dark hyperstained granular areas which were evident on NADH stain (x 200). J: Microphotograph showing muscle tissue with variation in fibre size and mild interstitial (endomyseal) fibrosis. One of the shows red granular material at the peripheral region (subsarcolemmal, arrow) [Modified Gomori Trichrome stain×200]. K,L: ATPase stain highlighting the variation in fibre size and increase in one type of fibre (Type II fibre predominance). [K -ATPas 9.4; L – ATPase 4.6]
Genetic results
We performed targeted gene panel (n = 53 genes) sequencing for detecting mutations in the coding region of the genes linked to LGMD and associated disorders. NGS analysis was done using the Agilent Sureselect QXT target enrichment kit (Agilent Technologies, Inc. Santa Clara, CA, USA) according to the manufacturer’s protocol. The enriched libraries were pooled and then subjected to sequencing on the Mi-Seq system (Illumina, Inc., San Diego, CA, USA). The patients from the three families (patients 1, 5, 6, and 7) showed identical homozygous missense mutation c.1000G > A (p.Asp334Asn) in the C-terminal region last exon 9 of the GMPPB gene (NM_021971.4). The remaining affected members in family 1 (patients 2, 3, and 4) were confirmed to have the same mutation by Sanger sequencing.
Muscle MRI findings
Independent histograms depicting the Mercuri scoring for the individual lower limb muscle are illustrated in Fig. 3. The Mercuri and Borsata scoring for individual lower limb muscles are enumerated in the Supplementary table.
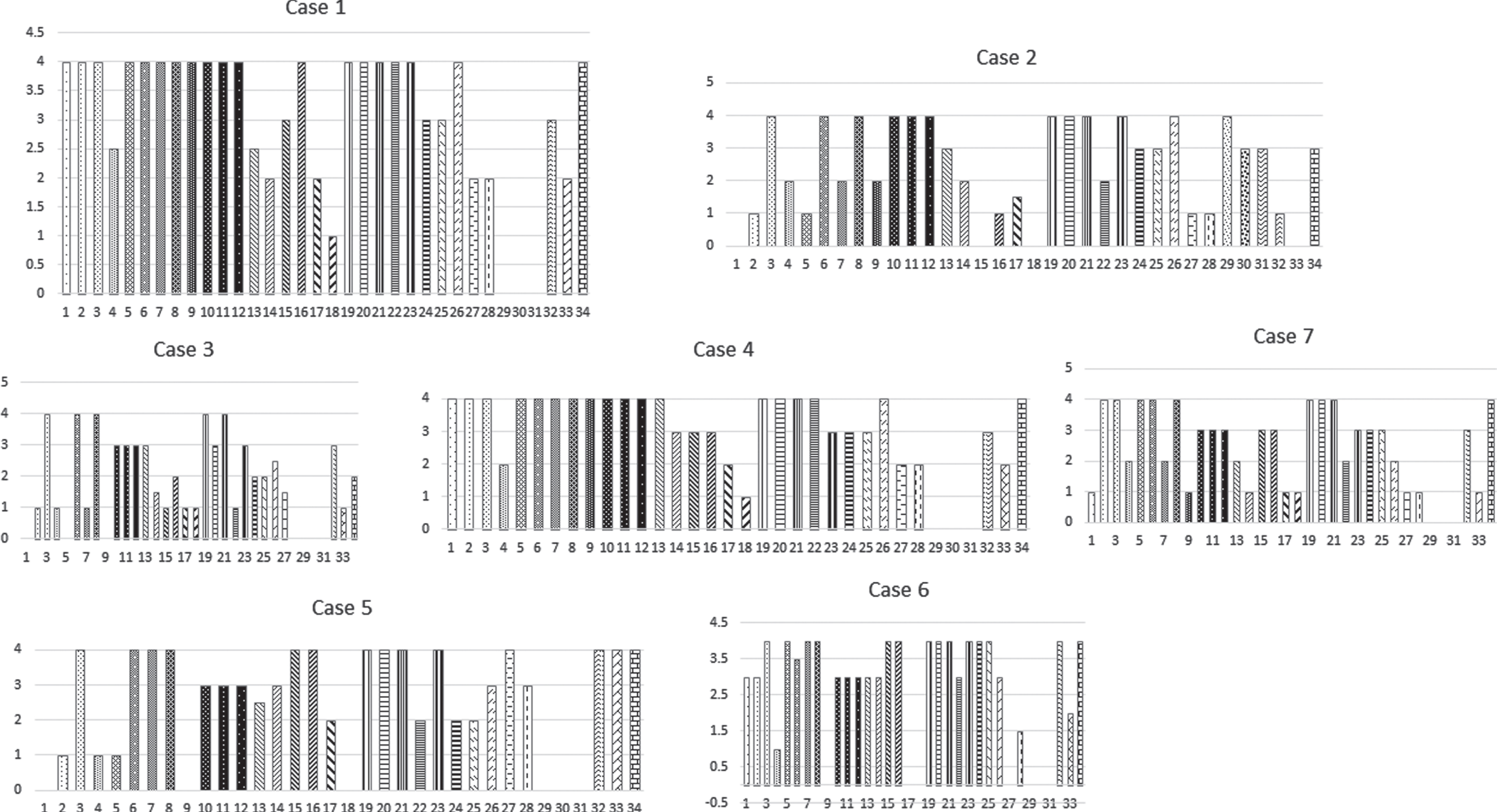
The individual histograms depict the Mercuri grade of fatty infiltration of the 34 assessedlower limb and paraspinal muscles of the individual patients. The proximal muscles are more affected correlating with the clinical severity. Muscles with normal power in legs also showed fatty infiltration.
Paraspinal and pelvis level
The paraspinal muscles at the lumbosacral level comprising erector spinae (longissimus dorsi, spinalis dorsi), the multifidus, and interspinales were involved uniformly as a group in all the patients. The mean T1 score (MTS) of this group was 3.4.
The Gluteus minimus and quadratus femoris demonstrated the maximum involvement (mean score = 4), followed by the adductors and obturators (externus = 3.9; internus = 3.2) (mean T1 score = > 3). Gluteus minimus and medius were more affected than gluteus maximus. Moderate involvement of gluteus medius and pyriformis (mean T1 score = 2.57) and mild involvement of tensor fascia lata (mean T1 score = 1.6) and pectineus (mean T1 score = <1.57) muscles were noted. There was a prominent proximo-distal / cranio-caudal gradient with more severe fatty infiltration within the proximal segment of the muscles when compared to distal parts (Figs. 4, 5).
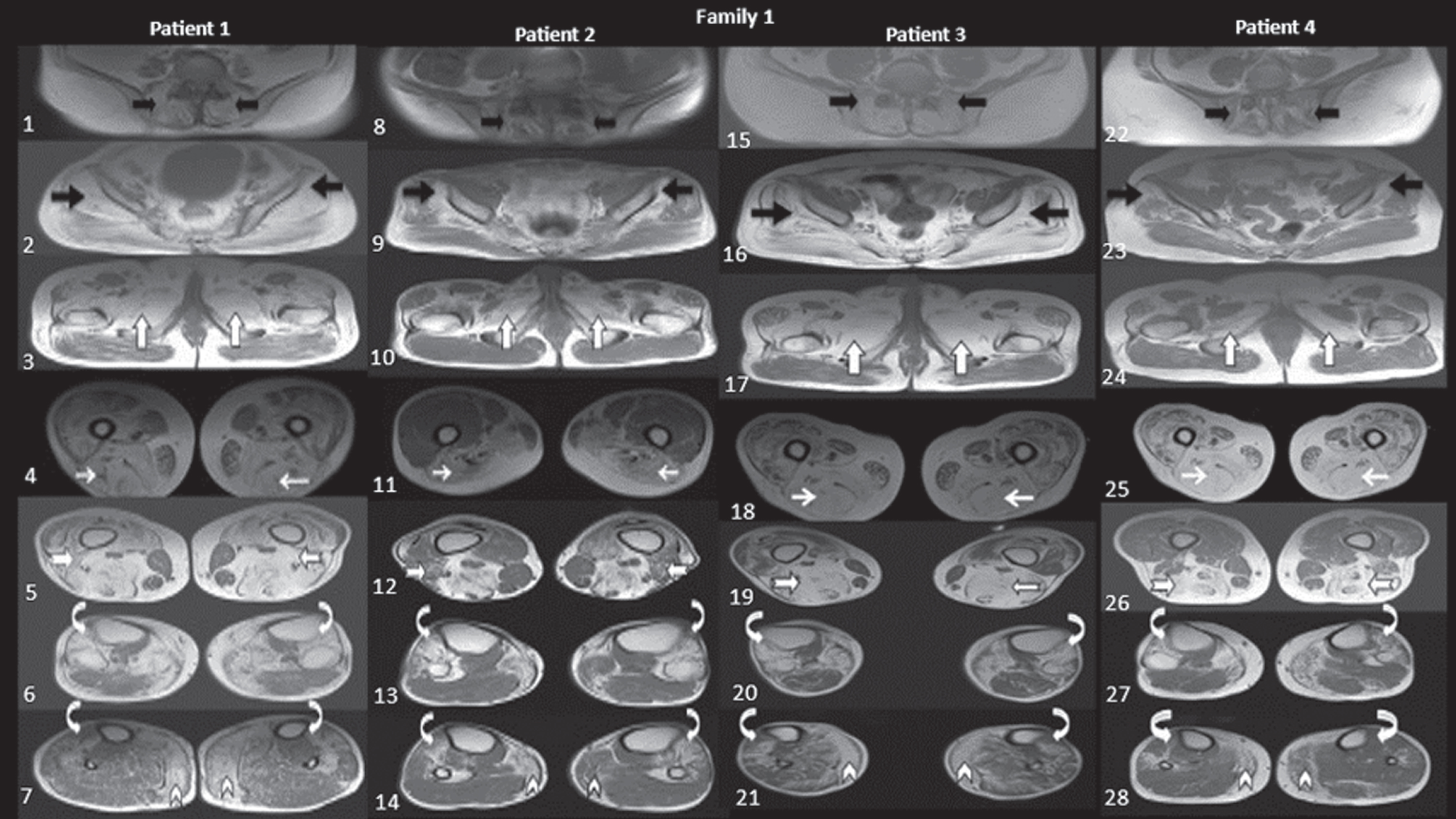
Family1:Patient 1(1–7), Patient 2(8–14), Patient 3(15–21), Patient 4(22–28), Muscle involvement is depicted in T1 axial images: First row (Fig. 1,8,15 and 22): Black arrows depict the severe paraspinal muscle involvement. Second row (Fig. 2,9,16 and 23): Black arrows shows severe gluteus minimus involvement, with varying severity of involvement of gluteus medius and spared distal aspect of gluteus maximus. Third row (Fig. 3,10,17 and 24): Upright white arrows depict the severely affected adductor group of muscles. Fourth row (Fig. 4,11,18 and 25): White arrows demonstrate severely involved hamstring muscles. Fifth row (Fig. 5,12,19 and 26): Notched white arrows demonstrate the spared short head of biceps femoris (biceps gradient) with severe involvement of other components of hamstrings, vastus medialis and intermedius involvement with relative sparing of the lateral aspect of vastus lateralis. Sixth row (Fig. 6,13,20 and 27): Curved arrows depict severely involved TA muscle groups with the involvement of extensors. Seventh row (Fig. 7,14,21 and 28): Chevron arrows depict the hypertrophied medial gastrocnemius muscle with fatty infiltration, also note the mediolateral gradient involvement in soleus muscles.
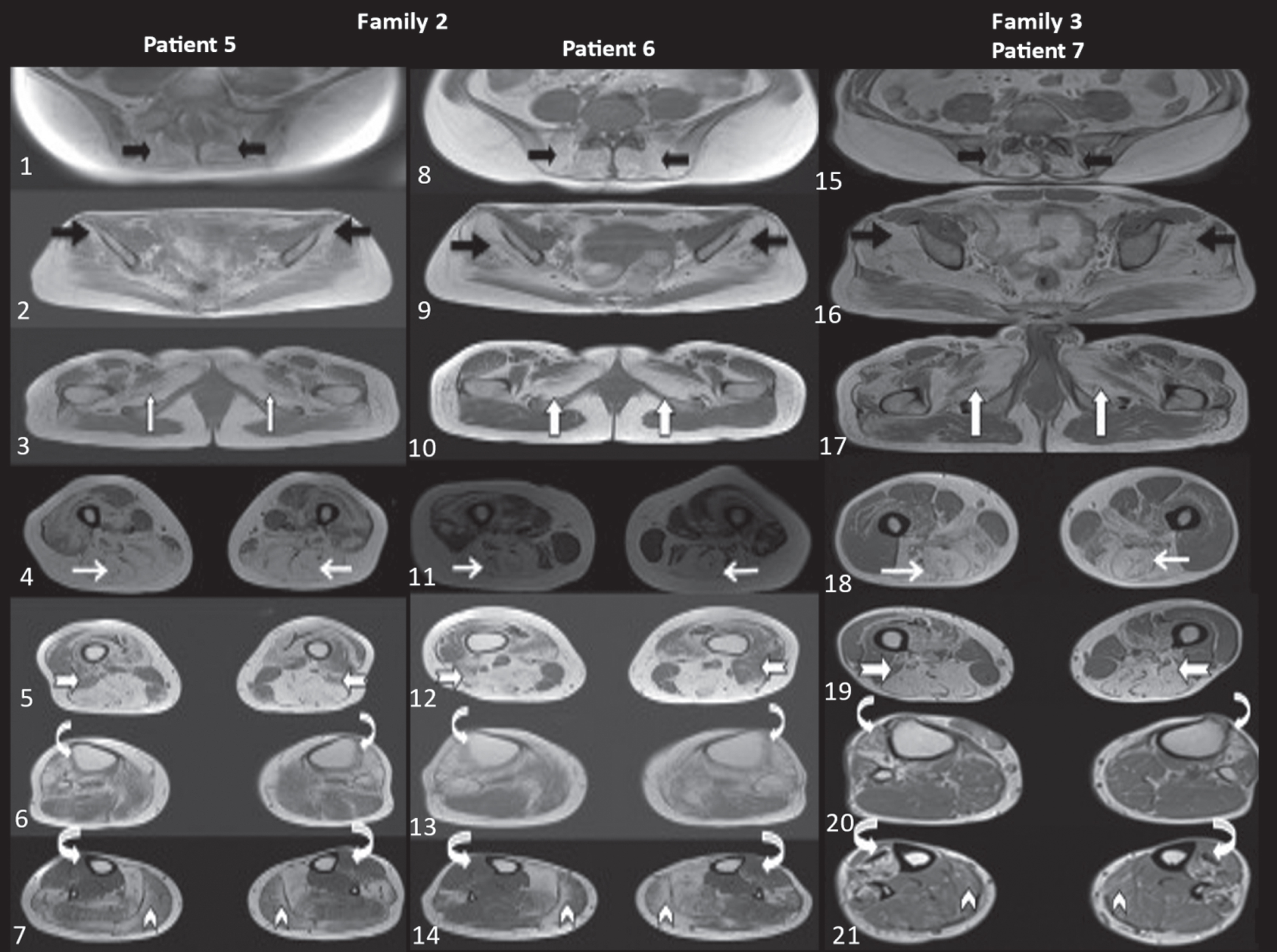
Family 2:Patient 5 (Fig: 1–7), Patient 6 (Fig: 8–14), Family 3: Patient 7 (Fig: 15–21), and controls-D (Fig: 22–28). Muscle involvement is depicted in T1 axial images:
At thigh level
The semimembranosus (SM), semitendinosus (ST), and long head of biceps femoris (LHBF) were most severely affected (means score = 4), while rectus femoris (RF) and vastus intermedius were moderately affected (mean T1scores = 2.85 and 3 respectively). Similar to pelvic muscles, a distinct proximo-distal/cranio-caudal gradient was conspicuous in vastii muscles with profound fatty infiltration in the proximal segments of the muscle when compared to the distal segments as we approach the insertion site. The lateral and posterior part of the vastus lateralis (VL) was spared despite severe involvement of the other parts of the muscle. There was preferentially less severe involvement (mean T1 score = 2.57) of the short head of the biceps, with a rim of the lateral aspect being spared. Sartorius was only mildly involved in four patients. Gracilis was involved in all patients with a preferential postero- anterior gradient except in patient 7 (Fig. 4, 1A).
At leg level
The medial head of the gastrocnemius muscle was hypertrophied and more severely involved than the lateral head. A mediolateral gradient was observed in the soleus, with the medial aspect being more severely affected. Similar to the thigh and pelvis, prominent involvement of TA was noted with a proximo-distal/cranio-caudal gradient (mean T1 score = 3.7), wherein a more severe fatty infiltration was apparent in the proximal parts of the muscle when compared to the distal part of the leg. The extensor digitorum (ED) and extensor hallucis longus (EHL) were affected in all patients, with severe involvement observed in five patients (Mercuri score = > 3). A striking peroneal gradient was noted, with peroneus brevis being more severely and frequently affected in all patients (mean T1 score = 3). These muscles also had a cranio-caudal gradient. There was sparing of the tibialis posterior (TP), flexor hallucis longus (FHL), and flexor digitorum longus (FDL) in all patients (Fig. 4, 5). Myoedema was noted in adductor longus (AL), VM, VL, gracilis, medial gastrocnemius, TA, peroneii, and FHL muscles (Table 2).
Literature Review and Comparison of major Studies on muscle MRI findings
Family 1
In family 1 variable age of onset was noted ext-ending from the first to fourth decade. An age-dependent gradient of involvement was evident. In patient 3, the paraspinals were more severely affected than glutei. There was increased involvement of the posteromedial thigh compartment with milder involvement of the short head of the biceps. In the legs, medial gastrocnemius was severely involved with early involvement of EDL and EHL and peronei (Fig. 4C). In the other 2 patients with advanced disease, there was diffuse involvement of all the muscles, including quadriceps,gracilis, and sartorius (patient 4), but with relative sparing of gluteus maximus (patient 2) (Fig. 4B). The RF, VI, and VM were more severely affected than VL. Postero-medial thigh involvement was appreciable. Soleus was spared in patient 3 despite advanced disease and mildly involved in patient 1. Asymmetrical involvement of a few pelvic muscles and VL was noted in patient 2 (Fig. 4B). Although by MRI staging, patients had advanced disease stage, distinct sparing of flexor compartment muscles of the leg was noteworthy in all patients. Further, this finding corroborated with the well preserved muscle power of plantar flexors. Myoedema was seen only in patient 4, who was the youngest with relatively early muscle involvement.
Family 2
Both patients had severe paraspinal muscle involvement, while patient 6 in the series also had profound involvement of the gluteus maximus. Both had significant involvement of anterior and posterior thigh muscles (Fig. 5A, B). Gracilis was mildly involved in patient 5. In the legs, medial gastrocnemius hypertrophy was noted, and there was severe involvement of the peroneii. Myoedema of VM and VI was noted in both the patients, while myoedema of gracilis was noted in patient 5 (Fig. 5A).
Family 3
Severe involvement of paraspinals, gluteus minimus, and maximus was noted at the pelvic level. More severe involvement of hamstrings and medial aspects of thigh muscles were noted. A definite cranio-caudal gradient of muscle involvement was noted in the gluteii and vastii. Mild and early involvement of gracilis and sartorious was also observed. Hypertrophy of the medial head of gastrocnemius was noted with severe involvement of anterior compartment muscles of leg and peroneus brevis. Myoedema was noted in the TA muscle (Fig. 5C).
The evolution of the pattern was different in the three families. There was almost similar severe involvement occurring in patients belonging to families 2 and 3 during the 4th decade compared to individuals from family 1 who were beyond the 5th decade. The involvement of gastrocnemius in the patient from family 3 was disproportionately milder when compared to other families. Peroneii were severely involved a decade earlier in family 2 (P5, P6) compared to family 1 (P1-P4). More muscles showed myoedema in family 2.
Correlation findings
Inter and intrafamilial variability: In family 1 the disease severity, disability, and duration of illness correlated with the severity of muscle involvement. In family 2 the proband (P5) was more severely affected as compared to the elder sibling (P6) with respect to the muscle weakness, disability, and MRI changes. Mild fatigable ptosis was evident in family 2 only. Also, with advancing age and increasing duration of illness, the MRI revealed more prominent and more extensive fatty infiltration among all the patients. The lone non-ambulatory patient (P1) and two siblings (P2, P3) requiring assistance to walk belonging to family 1 had the most severe involvement of the muscles on MRI. The women appeared to have more severe muscle involvement when compared to men with similar disease duration.
Algorithm for Muscle MRI based differential diagnosis
A simple algorithm for arriving at the possible diagnosis of LGMD-congenital limb-girdle myasthenic syndromes based on the pattern of lower limb muscle involvement on MRI has been derived (Fig. 6).
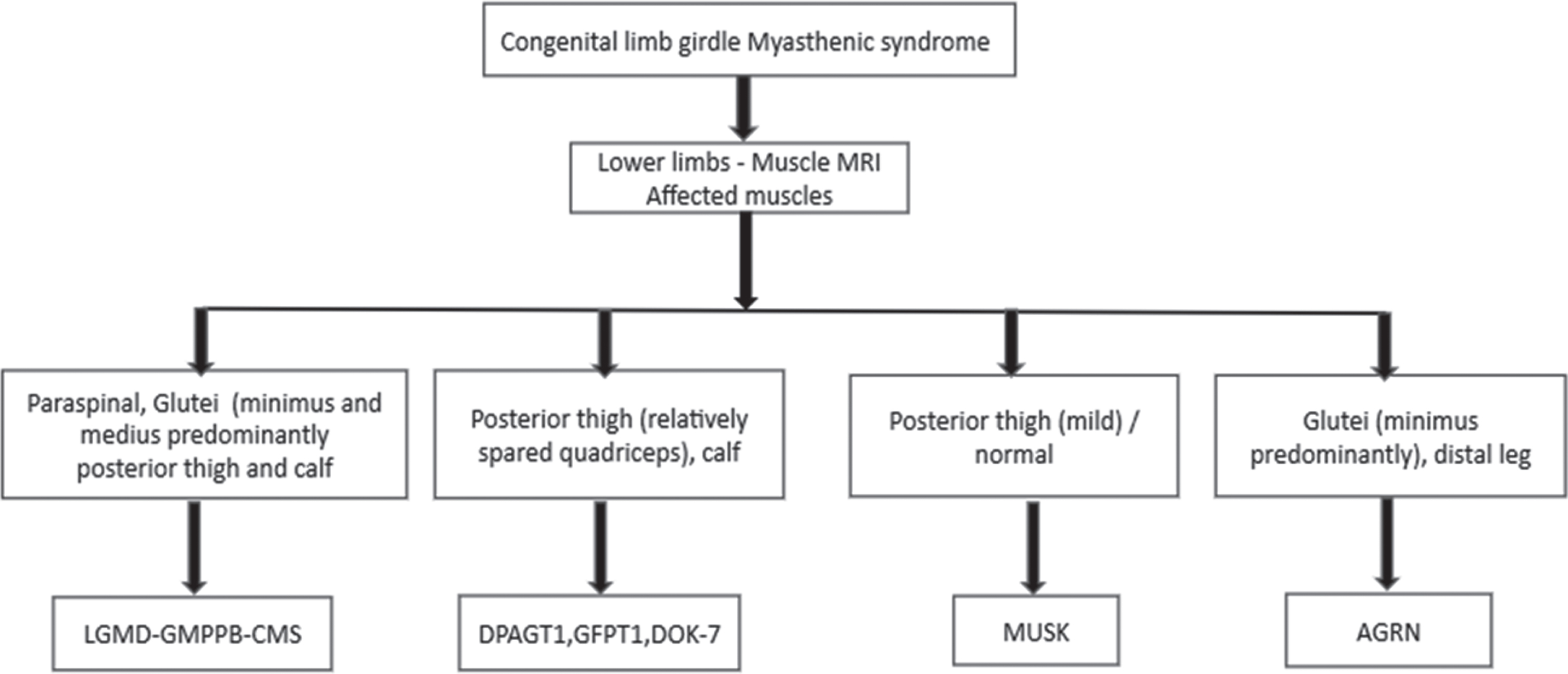
A simple algorithm for arriving at the possible diagnosis of congenital limb girdle myasthenic syndromes based on the pattern of lower limb muscle involvement on MRI.
DISCUSSION
The clinical phenotype of GMMPB related dystroglycanopathy varies from severe forms of CMD to milder forms of LGMD-CMS phenotypes [1, 5]. A clear history of fatigable weakness and fluctuating motor symptoms are vital clinical clues that suggest the diagnosis in these patients presenting with prominent LGMD phenotype. All our patients presented with a relatively late childhood phenotype to adult-onset gradually progressive fatigable limb-girdle weakness with moderate to severe truncal weakness. The patients with longer duration had more noticeable clinical features of primary muscle disease such as selective muscle weakness, calf hypertrophy, limb-girdle weakness with wasting, scapular winging, and hyperactive ankle jerks in almost all. All had elevated CK levels and showed prominent decrement response to RNS.
An identical homozygous missense mutation c.1000G > A (p.Asp334Asn) was identified in all our patients. This mutation has been previously des-cribed in compound heterozygous form in 2 patients of Pakistan and Indian origin. While muscle imm-unohistochemistry showed reduced glycosylated α-dystroglycan in both these patients, functional studies of the mutation from transfected myoblasts showed the formation of abnormal GMPPB protein aggregates in the cytoplasm [1]. However, in contrast to our patients who had milder limb-girdle CMS phenotype, they presented with severe forms of CMD without any features of CMS. This probably suggests a milder effect of c.1000G > A on GMPPB function when occurring in homozygous state rather than compound heterozygous state. Further, the c.1000G > A is reported only in the south Asian population (19 heterozygotes) as per gnomAD with a minor allele frequency (MAF) of 0.0006 and homozygotes were not present in the control population. The variant identified in our cohort is classified as likely pathogenic as per American College of Medical Genetics (ACMG) criteria [12]. Due to the presence of the identical south Asian specific variant in unrelated families and segregating in all affected family members, we suspected a possibility of founder effect and haplotype analysis was planned for these families.
The characteristic muscle MRI findings in our unique cohort showed prominent and severe involvement of all the lumbosacral paraspinal muscles, gluteus minimus, quadratus femoris, semimembranosus, semitendinosus, long head of biceps femoris, medial head of gastrocnemius, TA, extensor digitorum, extensor hallucis longus, and peroneus bre-vis. The moderately affected muscles were gluteus maximus and medius, hip adductors, obturatus, pyriformis, rectus femoris, vastus intermedius,gracilis, lateral head of the gastrocnemius, and medial part of soleus. The mildly affected muscles were tensor fascialata, pectineus, the short head of biceps femoris and sartorius (n-4/7). The muscles spared were: posterolateral part of VL, TP, FHL and FDL. Myoedema was prominently noted in adductor longus, VM, VL, gracilis, medial gastrocnemius, TA, peroneii, and FHL.
Similar to our findings, Oestergaard et al., have shown that the lumbar erector spinae muscles are the most severely involved, and the second most frequently affected muscle is the hamstrings, while the anterior thigh muscles were less affected. They also found an age-dependent gradient of muscle involvement, but there was no correlation between disease duration and severity of muscle involvement [3]. The pattern of early and severe involvement of the lumbar paraspinal muscles, hamstrings, and medial thigh muscle groups observed in our patients is similar to that described in other earlier reports with LGMD-GMPPB-CMS phenotype [7, 9]. Sun et al., in a single case of LGMD R19, performed MRI of the thighs and showed fatty infiltration of the medial compartment [13]. Luo et al., have shown selective involvement of calf muscles in one patient confirmed to have a compound heterozygous mutation in the GMPPB gene [6]. The preferential involvement of medial gastrocnemius with hypertrophy noted in our series is similar to the findings in previously reported studies [7, 14]. In the report by Montagnese et al., the ages at MR imaging were 74 and 23 years and their wholebody MRI had shown fatty replacement of paraspinal, thigh adductors, and calf muscles in the older patient and edematous changes of the soleus muscle in the younger patient.
Preferential posterior thigh muscle involvement is also known to be a feature of other abnormal glycosylation-related CMS phenotypes like GFTP1 and DPAGT1 [15, 16]. In addition, similar findings are noted in DOK-7 and MUSK-related CMS [17, 18]. However, these mutation-related CMS phenotypes have relative sparing of the quadriceps group of muscles, unlike observed in the LGMD-GMPPB-CMS phenotype. Nevertheless, Montagnese et al., have reported sparing of the quadriceps muscle. Unlike our cohort, preferential involvement of EDL and EHL has not been highlighted in any of the previous studies on LGMD-GMPPB-CMS patients, but a similar pattern of involvement of distal muscles of the leg is reported in AGRN related CMS [19].
The proximal segments were more severely affected, and a striking cranio-caudal/proximo-distal gradient was noted in the glutei, vastii, and peronei muscles with respect to fatty infiltration. A similar consistent finding of a proximo-distal gradient in vastii muscle is reported in sarcoglycanopathies [20]. Such a specific gradient is not described in previous studies on the GMPPB-CMS phenotype. The relative sparing of distal muscles with more severe involvement of proximal thigh muscles is consistent with the LGMD pattern of involvement, and its pathophysiology is yet to be elucidated [2, 3].
Surprisingly, sartorius and gracilis involvement was commonly identified in our patients with a discernable posterior to anterior gradient. In the report by Oestergaard et al., the MR images show the involvement of the sartorius. However, this is in significant variance with the findings demonstrated in the study group of Belaya et al., where in gracilis is spared and sartorius is spared in the report by Montagnese et al., [2, 7].
Furthermore, in two cases published by Cabrera-Serrano et al., the authors describe a 71-year-old Italian woman with GMPPB who had sparing of sartorius and gracilis even with end-stage involvement of all other muscles [8]. In contrast, all our patients, including those as early as in the third decade, had involvement of these two muscles. To the best of our knowledge, this pattern of involvement and gradients has not been documented before in the GMPPB-LGMD-CMS phenotype. Amongst the glutei muscles, preferential involvement of the gluteus minimus and medius over maximus was noted. A similar pattern of severe involvement is reported in CMS due to AGRNgene mutation [19], but not in GMPPB related muscle disease.
It was evident that even in the advanced stages of the illness, a rim of crescentic muscle tissue was noted in the medial gastrocnemius and short head of biceps femoris in our patients. Similarly, Oestergaard et al., and Cabrera-Serrano et al., have reported that the muscle involvement was not uniform with the progression of the disease [3, 8].
Sarkozy et al., have described the absence of muscle involvement on MRI in the mild phenotype of LGMD-GMPPB [21]. In contrast, the patients (P5,P6) from our family 2 had severe involvement of muscles at an earlier age.
Belaya et al., identified myoedema on STIR in gastrocnemius and soleus [2], while Montagnese et al., have described similar findings in soleus muscle only [7]. In contrast, myoedema was noted in AL, VM, VL gracilis, medial gastrocnemius, TA, peroneii,and FHL in our patients. The more extensive presence of myoedema in our patients is challenging to explain. Interestingly, myoedema in gastrocnemius, soleus, and distal leg muscles has been described in other CMS phenotypes related to AGRIN, GFPT1, and DPAGT1 [15, 16].
The MR imaging pattern observed inLGMD R19 GMPPB-related disorder) or its overlap with CMS has an association with other glycosylation-related disorders such as FKRP (LGMD2I) and POMT (LGMD 2N) [21, 22]. The preferential involvement of posterior thigh and calf muscles as seen in our patients are also observed in DPAGT1, GFPT1, and DOK-7.
Interestingly in patients with ANOC5 myopathy, WBMRI showed variable muscle involvement. Partly similar to our patients, the paraspinal, posterior thigh, and posterior leg muscles demonstrated asymmetric fatty replacement. There was patchy involvement of the anterior compartment muscles. In comparison, our patients with GMPPB-LGMD-CMShad symmetrical involvement of the muscles, with sparing of anterior/extensor compartment muscles and peroneal muscles. There was also involvement of TP and flexor compartment, which are relatively spared in our patients.
Furthermore, no proximo-distal gradient of muscle involvement was observed in their patients. Interestingly the female patients had a mild to asymptomatic course. However, in our cohort the female patients had a more severe manifestation in the clinical and imaging findings [23].
In an earlier study from China, muscle biopsy from two patients with compound heterozygous GMPPB mutations had shown findings of fibre size variation, hypercontracted fibre, and interstitial proliferation. There was a presence of protein aggregation in the cytoplasm attributed to the abnormal folding of GMPPB [6].
Our patients also showed similar findings on muscle biopsy. The histopathology studies on three of our cases showed variations, the histophenotypic spectrum in Patient 1 showing overt myopathic changes (dystrophic changes) while Patient 7 showed mild myopathic changes. Patient 5 demonstrated neurogenic changes in the muscle. These neurogenic changes could be part of the disease (pathobiology) process or secondary phenomenon, while myopathic features may not be evident in the samples tissue. Also, all these histological changes could be a part of the evolutionary process of the pathology where the full-blown pathology is not seen in the muscle biopsy submitted, and this prompts for more exploration into the natural biology/pathogenesis and pathophysiology of the disease. Unfortunately, we did not perform electron microscopy or western blot studies to confirm evidence of doubtful protein aggregates.
As patients with GMPPB related CMS are at least partially responsive to salbutamol and pyridostigmine, particularly in the early stages of the disease course, the imaging pattern described here can help in the early diagnosis of patients presenting with an LGMD like phenotype with fatigable weakness. In the current cohort, the younger patients with an early disease stage had a significant response to a combination of oral pyridostigmine (180 to 240 mg/day) and salbutamol (6 to 12 mg/day). The patients reported improvement in the stamina to perform physical activities and the muscle fatigability also improved. These improvements were not quantified.
The distinct muscle pattern may serve as a diagnostic clue for GMPPB-LGMD/CMS and seems to be somewhat consistent in a cohort of 7 patients with an identical homozygous variant with some inter and intrafamilial variability. Asymmetric muscle involvement is also observed in some patients. The limitation of our study is that we could not perform whole-body muscle MRI and electron microscopy studies on muscle tissue.
CONCLUSION
Hitherto, unreported, the pattern and gradient of muscle involvement identified in the current cohort are unique. Despite the presence of features of muscular dystrophy, the typical muscle involvement pattern coupled with the clinical history of easy fatigability and decremental RNS response will help in suggesting a possible diagnosis of GMPPB dystroglycanopathy. It could also help in differentiating other dystroglycan-related CMS and Limb-Girdle Myasthenic syndromes.It may be proposed that the unique muscle pattern may be a manifestation of the homozygous mutation of the GMMPB identified in this cohort. The limitations are the non-availability of muscle IHC / WB and EM studies. We also did not perform whole-body muscle MRI.
Footnotes
ACKNOWLEDGMENTS
The authors received no financial support for the research, authorship, and/or publication of this article. We would like to thank the patients and their families for participating in this study.
CONFLICT OF INTEREST
On behalf of all authors, the corresponding authors state that there is no conflict of interests.