
Abstract
Phenotypic cell-based high-throughput screenings play a central role in drug discovery and toxicology. The main tendency in cell screenings is the increase of the throughput and decrease of reaction volume in order to accelerate the experiments, reduce the costs, and enable screenings of rare cells. Conventionally, cell-based assays are performed in microtiter plates, which exist in 96- to 1536-wells formats and cannot be further miniaturized. In addition, performing screenings of suspension cells is associated with risk of losing cell content during the staining procedures and incompatibility with high-content microscopy. Here, we evaluate the Droplet-Microarray screening platform for culturing, screening, and imaging of suspension cells. We demonstrate pipetting-free cell seeding and proliferation of cells in individual droplets of 3–80 nL in volume. We developed a methodology to perform parallel treatment, staining, and fixation of suspension cells in individual droplets. Automated imaging of live suspension cells directly in the droplets combined with algorithms for pattern recognition for image analysis is demonstrated. We evaluated the developed methodology by performing a dose–response study with antineoplastic drugs. We believe that the DMA screening platform carries great potential to be adopted for broad spectrum of screenings of suspension cells.
Introduction
High-throughput phenotypic screenings of live cells play a pivotal role in the fields of drug discovery and toxicology. Across all therapeutic areas, 32% of drugs approved between 1999 and 2008 were discovered using phenotype-based approaches. 1 Phenotypic screenings are also used as an in vitro model for toxicity tests and for studies of drug-induced liver injury (DILI).2,3 The constantly growing need for new drugs and bioactive compounds, the creation of large chemical libraries, and the development of sophisticated biological assays have resulted in a constant growth of market for cell-based assays, which was already estimated to be about US$11 billion in 2015. 4
Conventionally, cell-based assays are performed in microtiter plates, ranging from the 96-to 1536-well format. The tendency for increasing the throughput and decreasing the reaction volumes arises from the purpose of reducing the costs of experiments and enabling the screening of rare cells. The majority of spending, up to 75%, on high-throughput screenings (HTSs) goes toward reagents, microtiter plates, and assay kits. 5 Decreasing the volume per experiment combined with the increase in throughput is a way to reduce the total costs of the HTS experiments. On the other hand, more researchers are switching to human primary and stem cells for their cell-based screening. Such cells are often available in limited amounts and cannot be easily expanded, which makes it difficult to perform large screens. Miniaturization can solve this problem by decreasing the amount of cells per experiment, allowing for more compounds to be screened on rare cell types. Miniaturization of microtiter plates has reached its physical limit as further reduction of volumes of wells is difficult due to capillary effects. 6 At the same time, there is a clear need for further miniaturization in big pharmaceutical companies. 7
Miniaturization of assay format by means of alternative technologies has been a trend for some years now. Such technologies are represented by cell microarrays introduced by Sabatini and colleagues and microfluidic platforms of different kinds adopted for cell-based applications.8–12 Cell microarray technology, which is based on preprinting transfection mixtures onto a glass substrate prior to seeding a layer of adherent cells onto it, was widely used by multiple academic groups to perform phenotypic screens.13–16 Cell-based assays in a microfluidic format, where liquids are confined in small channels and cavities, can benefit from low sample consumption, high-throughput, precise and rapid addition of defined solutions, and reduced time of experiment and analysis. 17 However, along with mentioned advantages, these technologies have limitations. Cell microarrays are (1) limited to adherent cell cultures and not compatible with cells of a suspension nature, (2) designed for transfection assays and barely compatible with compound screenings, and (3) suffering from the risk of cross-contamination between the spots. Cell assays in microfluidic formats are proved to be advantageous compared with conventional method in such areas as single-cell sequencing, metabolism studies, ion channel research, and chemotaxis. 17 At the same time, microfluidics does not offer optimal solutions in multiple areas, including toxicity testing or high-content screenings. Therefore, such technologies are still restricted in their applications and cannot substitute microtiter plates for high-throughput cell screens in big pharmaceutical companies.
The screening of suspension cells as a rule is more challenging than the screening of cells of an adherent nature. This is first due to potential loss of cells during washing, staining, and fixation procedures. Second, suspension cells are small and tend to clump while growing, which makes it difficult or impossible to perform microscopy and image analysis. 18 In addition, suspension cells are not growing in one focal plane, being rather distributed throughout the whole volume. In order to perform microscopy of suspension cells, they are usually fixed first, making them not compatible with live imaging. Alternative miniaturized screening platforms are not always compatible with the screening of suspension cells due to the same reasons. For example, it is challenging to create cell microarrays with suspension cells, because cells would have to be immobilized on the surface using anchor molecules, which might affect the cell behavior and transfection efficacy. 19
Recently, we developed a Droplet-Microarray (DMA) platform for cell screenings and have been continuously optimizing different aspects of this technology.20–25 The DMA technology is based on superhydrophilic (SL)-superhydrophobic (SH) patterning. The effect of discontinuous dewetting enables formation of arrays of droplets trapped in SL areas. Biocompatibility of the polymer surface allowed us to adopt such arrays for cell culturing and create arrays of droplets containing cells. The unique feature of the DMA platform is the ability to form homogeneous droplet arrays spontaneously without a need for pipetting of each droplet. We demonstrated the compatibility of the DMA technology with culturing of different cell types in volumes ranging from 3 to 80 nL. 21 We established different methods to deliver compounds and reagents into droplets containing cells and demonstrated the applicability of such methods for drug treatment and transfection of adherent cells. In the current study, we further exploited the potential of the DMA platform and developed protocols for the culturing and screening of suspension cells.
In this study, we developed a simple methodology to perform staining and fixation of suspension cells in individual miniaturized droplets in a parallel pipetting-free manner without losing the content of the droplets. We designed and manufactured a handheld device, which enables easy manipulations with cells. We also developed a semiautomated workflow for high-throughput compound screening of cells on the DMA platform based on microscopy of live cells and demonstrated its applicability by testing anticancer compounds. We believe that due to its unique features, the DMA screening platform in combination with methodology developed in this work, carries the potential to be widely adapted by other researchers for phenotypic screenings of suspension cells.
Materials and Methods
Preparation of DMA Slides
Preparation of SL-SH patterned surface of DMA slides was described in detail elsewhere.21,26 Briefly, patterns were prepared as follows. Glass slides (Schott Nexterion, Jena, Germany) were first activated with 1 M NaOH (Carl Roth GmbH + Co. KG, Karsruhe, Germany) for 1 h, followed by neutralization with 1 M HCl (Merck KGaA, Darmstadt, Germany) for 30 min. Afterward, activated glass slides were modified with 20% v/v solution of 3-(trimethoxysilyl)propyl methacrylate (Sigma-Aldrich Chemie, Munich, Germany) in ethanol for 30 min at room temperature. A polymer layer was formed by applying 25 μL of polymerization mixture (24 wt% 2-hydroxyethyl methacrylate [HEMA] [Sigma-Aldrich Chemie], 16 wt% ethylene dimethacrylate [EDMA] [Sigma-Aldrich Chemie], 12 wt% 1-decanol, 48 wt% cyclohexanol [Sigma-Aldrich Chemie], and 0.4 wt% 2.2-dimethoxy-2-phenylacetophenone [Sigma-Aldrich]) onto a fluorinated glass slide, covering it with a modified glass slide and cross-linking the polymer by ultraviolet (UV) irradiation with 12 mW/cm2 intensity and 260 nm wavelength for 15 min. Glass slides were fluorinated in trichloro(1H,1H,2H,2H-perfluorooctyl)silane (Sigma-Aldrich Chemie) vapor in desiccators under 50 mbar vacuum overnight. The polymer surface was modified with alkyne groups by incubating the slides in solution containing 45 mL of dichloromethane (Merck KGaA), 56 mg of 4-(dimethylamino)pyridine (Novabiochem, Merck KGaA), 111.6 mg of pentynoic acid (Sigma-Aldrich Chemie), and 180 µL of N,N′-diisopropylcarbodiimine (Alfa Aesar GmbH & Co KG, Karlsruhe, Germany) for 4 h under stirring at room temperature. The SH background was created by applying 5% v/v solution of 1H,1H,2H,2H-perfluorodecanethiol (Sigma-Aldrich Chemie) in acetone onto the polymer surface and irradiating the slide through a photomask (Rose fotomasken, Bergisch Gladbach, Germany) with 260 nm of UV light at 12 mW/cm2 for 1 min. Afterward, SL spots were created by applying 10% v/v β-mercaptoethanol (Alfa Aesar) solution in 1:1 water–ethanol onto the patterned surface and irradiating the slide with 260 nm of UV light at 12 mW/cm2 for 1 min.
Cell Culture
Jurkat human T-cell lymphocyte cells were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS, Sigma-Aldrich) and 1% penicillin/streptomycin (Gibco, Life Technologies GmbH, Darmstadt, Germany). Cells were cultured in T25 flasks and diluted every 2–3 days until a cell density of 2 × 105 cells/mL was achieved.
DMA slides for culturing were sterilized in ethanol and preconditioned by immersing in RPMI-1640 medium supplemented with 1% heat-inactivated FBS and 1% penicillin/streptomycin for 45 min in a cell culture incubator. Afterward, DMA slides were dried for 45 min under a clean bench. Cells were seeded onto a preconditioned DMA slide using a well-established protocol described in a previous work.
21
Briefly, 1.7 mL of cell suspension was applied per field surrounded by a SH border (
Fig. 1a
,
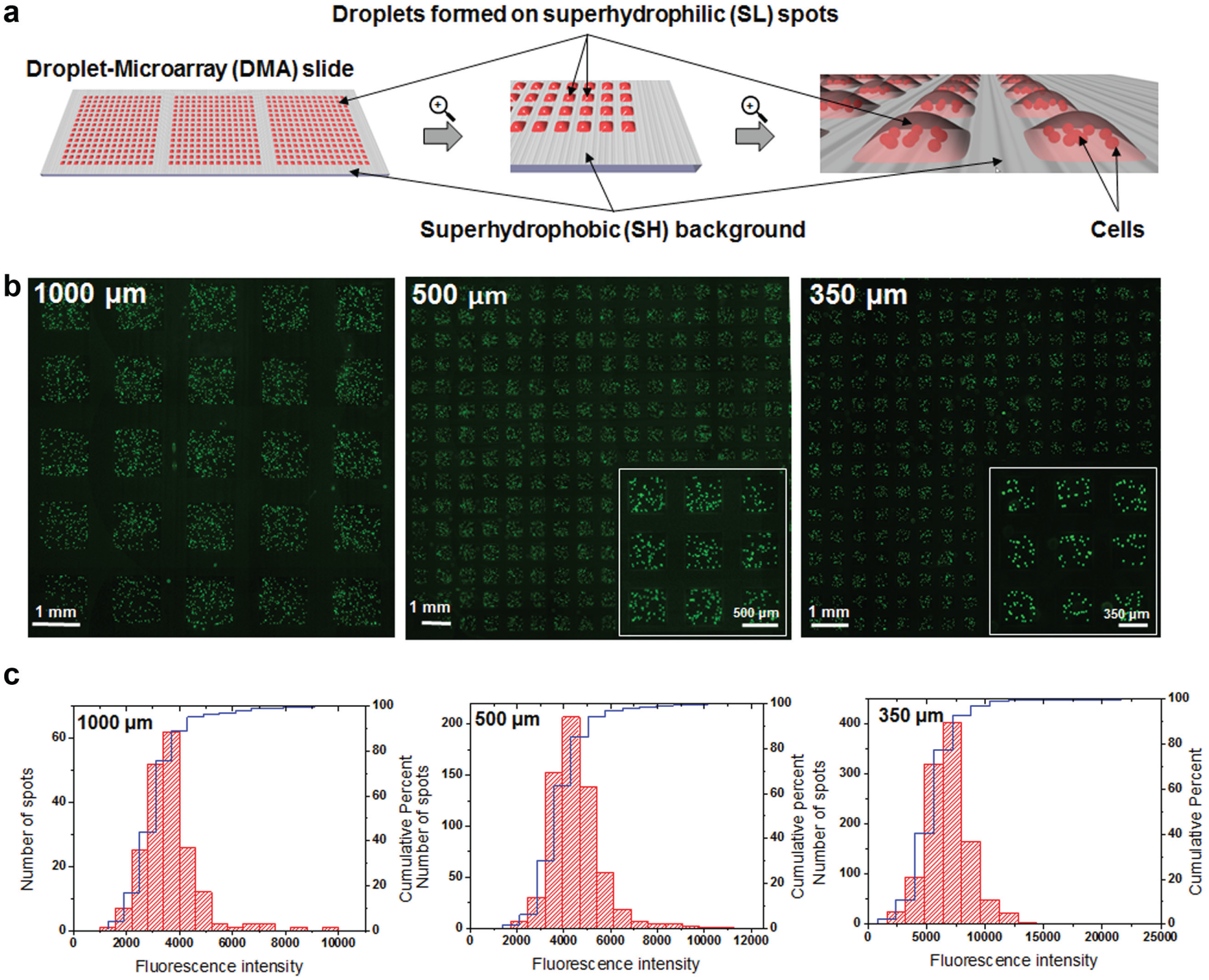
Culturing and microscopic analysis of suspension cells on the DMA platform. (
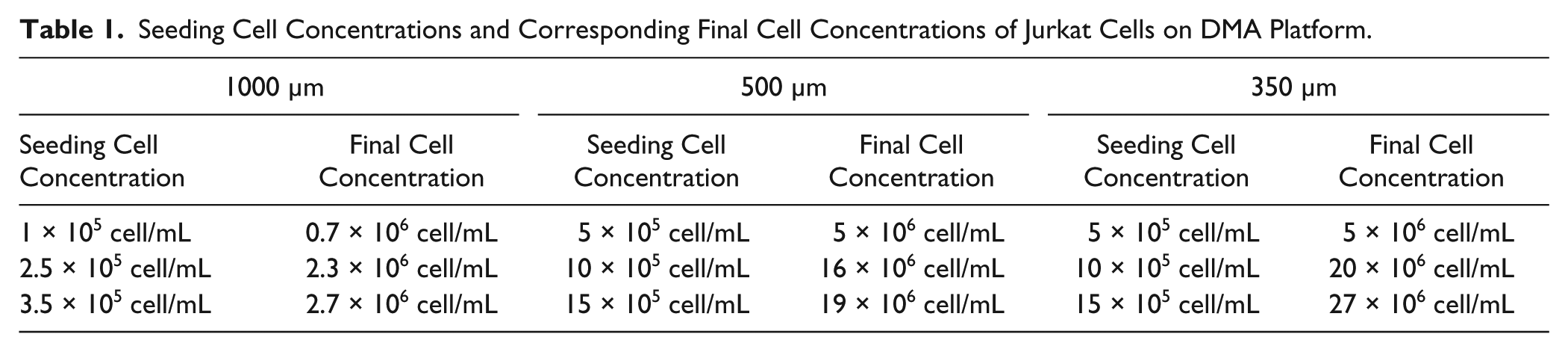
Seeding Cell Concentrations and Corresponding Final Cell Concentrations of Jurkat Cells on DMA Platform.
Cell Growth and Viability Study
Jurkat cells were seeded onto DMA slides with spots of different sizes and the amount of cells was estimated by monitoring 10–20 spots per slide at 0, 24, 48, 72, and 96 hours after seeding. Images of each spot were taken at 10× resolution using a Keyence BZ-9000 microscope (KEYENCE, Osaka, Japan). The amount of cells was estimated by manual counting. The viability of cells was estimated at a final time point at 96 h by live/dead staining (see Live/Dead Staining of Cells section). The data from three independent experiments were included in the analysis.
Aligning Frame for Sandwiching
An aligning frame was designed and manufactured for the aligning and sandwiching of DMA slides. The following parameters were taken into consideration: (1) secure holding of the slides; (2) precise (nonadjustable) alignment in the XY plane of both slides, with respect to each other; (3) a precisely adjustable distance of 0–2 mm between the slides in the z axis (sandwiching); and (4) the distance between the bottom surface of the frame and the lower DMA slide does not exceed 1 mm for direct microscopy in the frame. The holder consists of two separate frame pieces, an upper and lower holder. The slides are securely held in place via spring-loaded brackets. Both frames are precisely aligned in the XY plane, with respect to each other. This is achieved by four stainless steel pins, located in the four corners of the lower holder. These pins are aligned with four mirroring holes on the upper frame. Four holes contain polytetrafluoroethylene (PTFE) bushings (GGB Heilbronn GmbH, Heilbronn, Germany). The precise and adjustable distance between the slides is achieved by four micrometer screws (Lehmann Präzision GmbH, Hardt, Germany). These are placed symmetrically in the upper holder while being supported by the lower holder. The distance between DMA slides is adjusted by rotation of the screws. The holders and brackets were machined out of aluminum (Maschinenbau Kaltenbach GmbH, Crailsheim, Germany) and black anodized (Grau & Wagenblast, Abtsgmünd, Germany).
Sandwiching Method
The sandwiching method was used for all manipulations with individual droplets on DMA, including staining and fixation of cells and compound addition. The DMA slide containing cells was fixed in the lower holder of the aligner. For staining and fixation, an empty DMA slide was fixed in the upper holder of the aligner and fixation or staining solution was spread onto a DMA slide using an automatic pipette (
Fixation of Cells
The fixation solution consisted of 7.4% formaldehyde and 50 µg/mL DAPI in Dulbecco’s PBS. Fixation of cells was performed using the sandwiching approach as described above for 15 min on a bench. Afterward, the aligning frame was opened and the DMA slide containing cells was dried on air and then mounted with Immu-Mount (Thermo Fisher Scientific Inc., Darmstadt, Germany) and dried at +4 °C. Images of each individual droplet were taken using a Keyence BZ-9000 microscope (KEYENCE).
Live/Dead Staining of Cells
The staining solution consisted of 50 µg/mL propidium iodide (PI) (Invitrogene, Merelbeke, Belgium) and 50 µg/mL CalceinAM (Thermo Scientific) in PBS containing CaCl2 and MgCl2. Live/dead staining was performed using the sandwiching approach as described above for 15 min in a cell culture incubator. Afterward, the aligner was opened and the DMA slide containing stained cells was placed into a four-well dish (Thermo Scientific Nunc) and immediately subjected to microscopic analysis. Depending on the procedure, images were taken using either a Keyence BZ-9000 microscope (KEYENCE) or automated screening Olympus IX81 microscope (Olympus, Tokyo, Japan) (for details, see Automated Imaging and Image Analysis section).
Screening Initiation
Printing of chemical compounds onto glass slides was performed as follows. Clean microscope glass slides (Schott Nexterion) were fluorinated by trichloro(1H,1H,2H,2H-perfluorooctyl)silane (Sigma-Aldrich) under 50 mbar vacuum in a sealed desiccator containing an open vial of the silane overnight. Afterward, different amounts of antineoplastic drugs (in DMSO or diluted in pure RPMI-1640 medium if necessary) were printed on fluorinated slides in a defined pattern geometry corresponding to the geometry of DMA slides using the sciFLEXARRAYER S11 liquid dispenser (Scienion, Berlin, Germany). The following antineoplastic drugs were used: doxorubicin (European Pharmacopolia Reference Standards, European Directorate for the Quality of Medicines & Healthcare, Strasbourg, France), daunorubicin, cytarabine, dexamethasone, vincristine, and etoposide (Sigma-Aldrich). Prior to use, slides containing drugs were dried in a sealed desiccator containing silica gel (Sigma-Aldrich) for 24 h in darkness.
Jurkat cells were seeded onto a DMA slide as described in the Cell Culture section 3 h prior to screening initiation. Screening was initiated by sandwiching the DMA slide containing cells with the slide containing preprinted drugs as described in the Sandwiching Method section for 15 min in cell culture incubator. Afterward, the frame was open and the DMA slide containing cells was returned to the cell culture incubator.
Automated Imaging and Image Analysis
The DMA slide containing cells was placed into a four-well dish (Thermo Scientific Nunc), which was fixed in a standard holder for microtiter plates for microscopy. Imaging was performed using the automated screening microscope Olympus IX81 (Olympus). The grid of the DMA pattern was defined in the microscope software. The autofocus function “interpolate AF” was used to minimize defects that can arise from uneven surface thickness and unparalleled slide position. Z stacks of 10 slices were made to count for suspension cells being on slightly different focal planes. The images of one central field of view per each SL spot were taken at 10× magnification, in three different channels (brighfield, mCherry [for PI staining], and green fluorescent protein [GFP, for CalceinAM staining]). Imaging of 100 SL spots took about 20 min and resulted in 7.67 GB of data in total.
For analysis, images were processed using the “Extended Depth” algorithm in ImageJ software. To estimate the number of calcein- and PI-stained cells, a special algorithm for pattern recognition was developed. Obtained images were 12-bit greyscale images with a dynamic range of 4096 brightness values. In each image, the minimum brightness was calculated and brightness exceeding a fourth of the dynamic range was defined as the signal threshold. Each GFP and PI image was processed by applying normalization, shading correction, Laplacian of Gaussian filtering, noise cancellation, and application of a watershed transform, resulting in images showing detected objects. All found objects were counted if the maximum brightness value overcame the above-mentioned threshold. Each counted object was assigned the features area and mean intensity. GFP-positive cells tend to form clusters where individual cells cannot be identified automatically. To account for this, we assumed that a regular cell consists of 200 pixels and divided the area of a cluster by this number, getting an estimation of the number of cells within the cluster. To account for dead cells that exhibit a low GFP signal, every object found to be positive in the GFP channel was double-checked in the PI channel, and excluded from the count if found to be PI positive. All found objects were counted in both channels, delivering an estimation of objects in both GFP and PI channels.
Statistics and Data Analysis
Every experiment was repeated at least three times, and the data from three independent experiments were included in the analysis. Compounds in each concentration and appropriate controls were spotted in 10 repeats. The dose–response curves of tested drugs were plotted in GraphPad Prism 7. The sigmoidal curves were created based on a nonlinear regression technique. IC50 values of the compounds were calculated in the program.
The Z′ factor was calculated as follows:
Results
Suspension Cells on Droplet-Microarray: Culturing and Manipulation
We chose a Jurkat T-cell leukemia suspension cell line as a model for validating all the procedures that can be performed using the DMA platform. The DMA platform was introduced in a previous work.21,22 It consists of an array of SL spots on an SH background (
Fig. 1a
). Due to the extreme difference in wettability between SL and SH areas, arrays of droplets containing cells can be created by spreading the cell suspension on the surface of a DMA slide without the need to pipette each individual spot (
Fig. 1a
,
b
,
As a next step, we monitored viability and proliferation of Jurkat cells cultured on a DMA slide for 5 days without medium exchange. As demonstrated in
Figure 2b
and
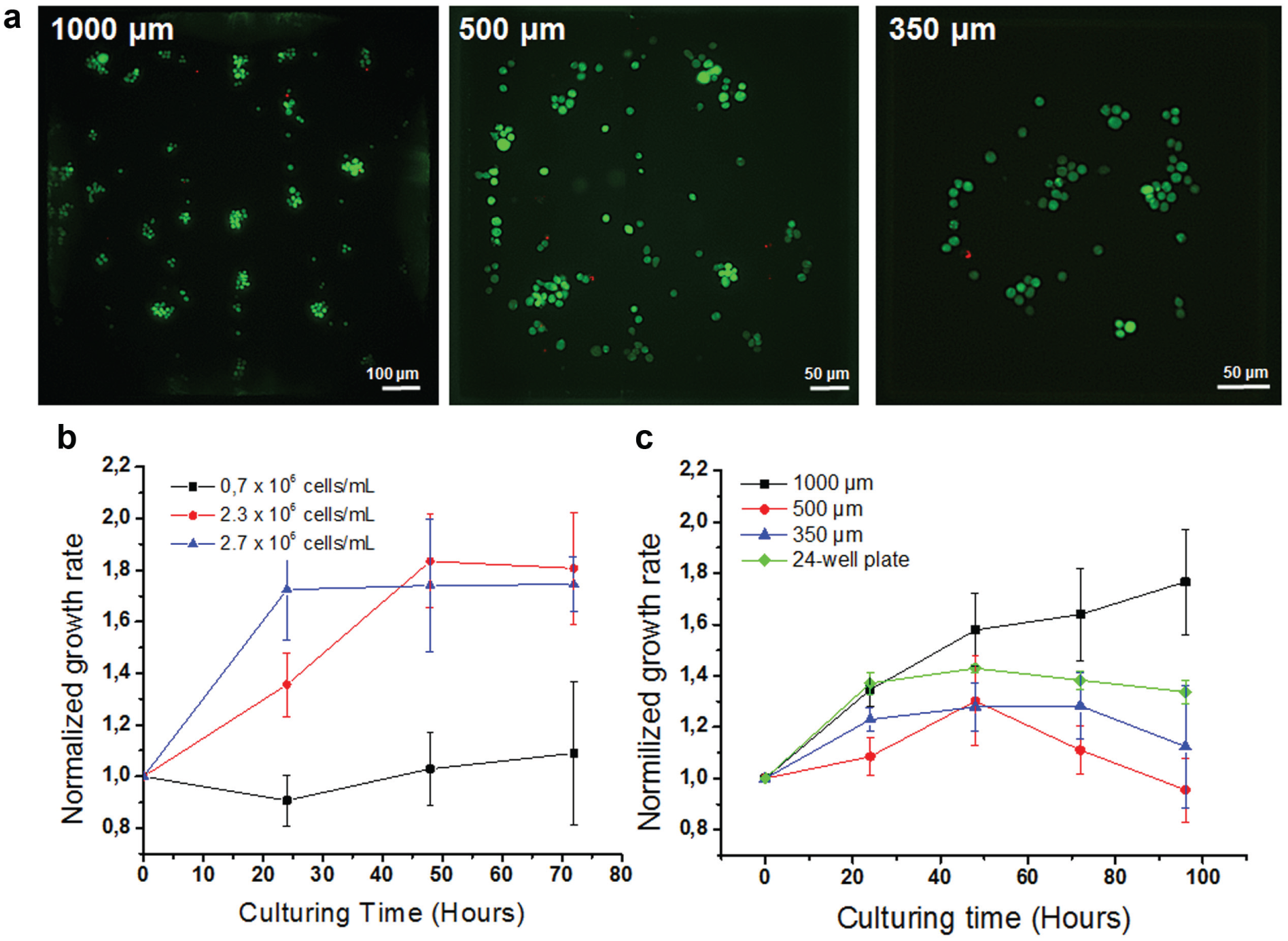
Viability and growth rate of Jurkat cells cultured on DMA slides with different spot sizes. (
Screening of suspension cells is challenging due to the risk of losing the cell content during washing and staining procedures. Therefore, in order to be able to use the DMA platform for screening applications, it is necessary to develop protocols enabling the addressing of each individual spot without disturbing the content of the droplets. Therefore, we validated the sandwiching method, introduced in a previous work,
22
for performing parallel staining and fixation of suspension cells. The principle of the sandwiching method for staining and fixation of cells on DMA is schematically presented in
Figure 3a
. The staining or fixation solution was spread onto a DMA slide with a pipette taking advantage of the effect of discontinuous dewetting (

HTS of suspension cells using the DMA aligning frame. (
High-Throughput Screening Using the DMA Platform
To be able to use the DMA platform for HTS applications, it is necessary to be able to perform experiments in a parallel manner. For this purpose, we developed a workflow for high-throughput drug screening of suspension cells on the DMA platform depicted in
Figure 4
. For a proof-of-concept study, we used DMA slides containing 588 spots of 1000 µm size (
Figs. 1a
and
4(2)
). In this workflow, drugs in different concentrations, including appropriate controls, were printed onto a hydrophobic glass slide in geometry corresponding to the geometry of a DMA slide and dried (
Fig. 4(1)
). Jurkat cells were seeded onto a DMA slide using a protocol established in previous work
21
(see Materials and Methods). This protocol allows for parallel pipetting-free seeding of cells on the full DMA slide using the effect of discontinuous dewetting (
Fig. 4(2)
,
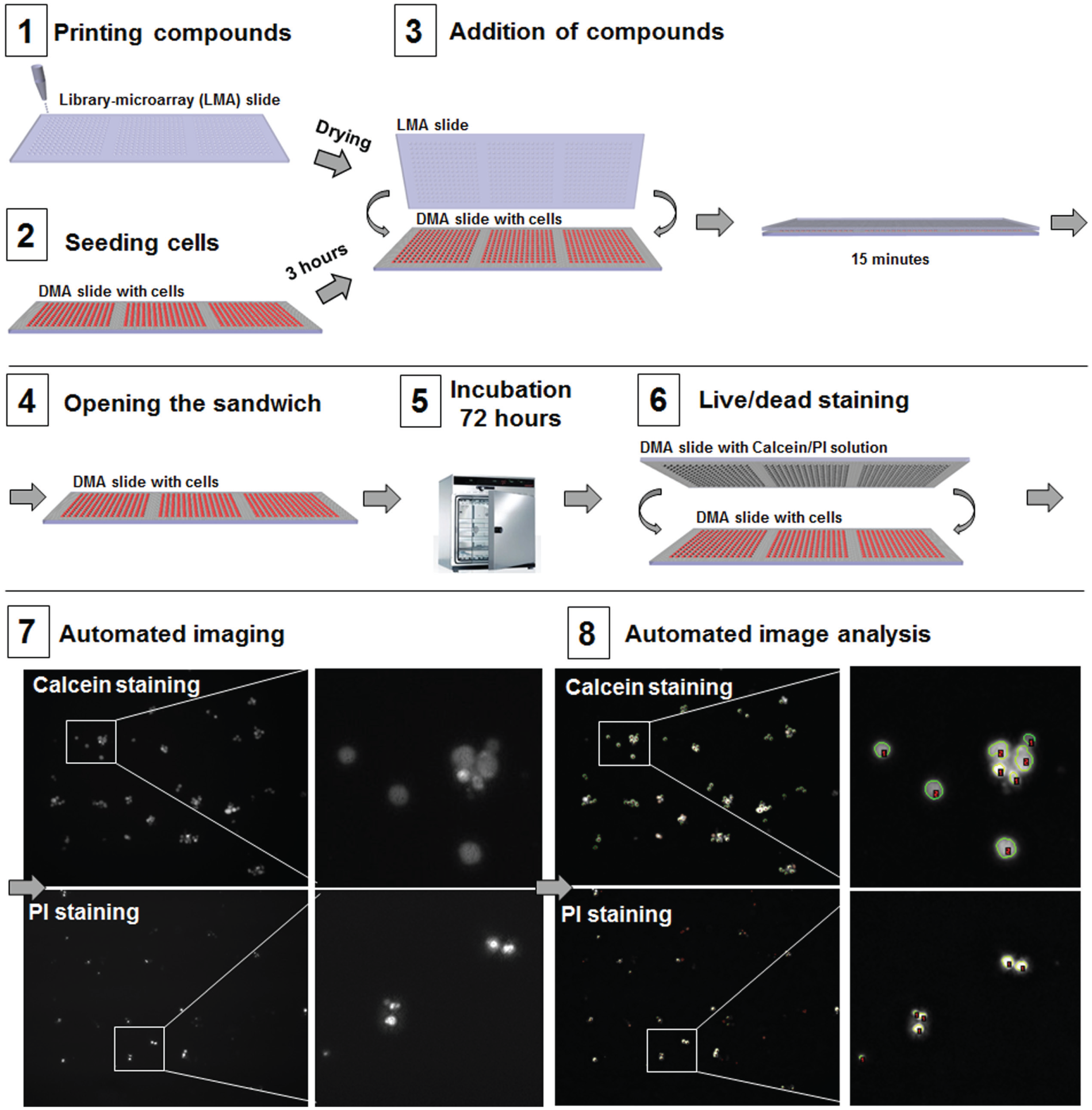
The workflow of HTS of suspension cells using the DMA platform.
Drug Treatment: Proof-of-Concept Study
To validate the developed protocol, we performed a proof-of-concept study where we tested Jurkat cells against antineoplastic drugs commonly used for the treatment of leukemia patients in the clinic and known to be effective against Jurkat cells (
Fig. 5
, Fig. S2). Using the workflow described above, we introduced Jurkat cells to different concentrations of the drugs. Cells were seeded onto DMA slides as described in Materials and Methods and placed in a cell culture incubator for 3 h before the screening initiation. Afterward, cells were introduced to 10 different concentrations of drugs and appropriate controls with 10 repeats for each concentration. Cells were incubated for 72 h, and the effect of drugs was estimated based on the viability of cells in treated spots compared with the control of untreated spots. Simultaneous live/dead staining of the cells, automated imaging, and image analysis were performed as described in the screening workflow. The obtained dose–response curves are demonstrated in
Figure 5
and
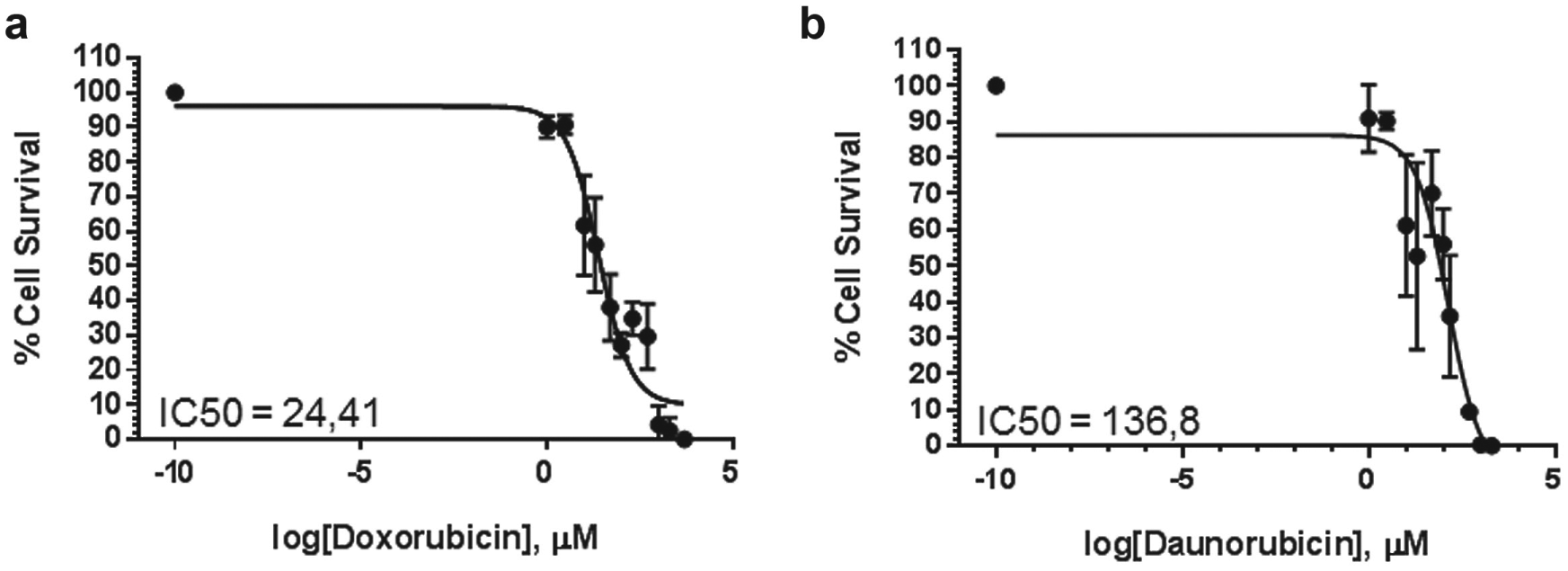
Drug treatment: Proof-of-concept study. Dose–response curves of doxorubicin (
Discussion
In the current study, we presented a system for HTS of suspension cells based on the DMA technology. DMA slides used in this work enable miniaturized screenings of live cells in droplets ranging from 3 to 80 nL in densities from 588 to 4563 spots per standard microscope glass slide, which corresponds to approximately 6000 and 50,000 spots per area of a standard microtiter plate, respectively. We utilized a previously established seeding method based on pipetting-free spreading of the cells on the whole DMA slide, taking advantage of the effect of discontinuous dewetting.21,22 Homogeneous distribution of suspension cells between the droplets of different sizes was demonstrated. The single-step pipetting-free seeding results in savings in the pipetting steps, pipetting tips, robotics, and time of the experiment.
We demonstrated that Jurkat cells continuously proliferate in individual droplets of 80 nL volume for at least 5 days without medium exchange. This makes the DMA platform compatible with assays performed within several days, which are the majority of primary screens, including such screenings as small-compound screenings and genome-wide siRNA screens.28–31 Cell proliferation itself is a critical parameter in many assays, for example, in testing of antineoplastic drugs that affect rapidly proliferating cells and might not be effective on slowly or nondividing cells.32,33
We developed a simple methodology based on a sandwiching approach for parallel addressability of individual droplets containing cells. The steps of compound addition, staining, and fixation of cells are performed in a one-step simultaneous procedure on the whole DMA slide. Such parallelization could be beneficial in the case of very short incubation times or rapid cell responses, where seconds or minutes are critical for the accurate readout. Since the sandwiching approach does not require multiple pipetting steps, the processing time for hundreds of “microwells” reduces to a few minutes of hands-on time. Finally, the DMA platform, in combination with the sandwiching method, can potentially enable affordable HTSs without a need for any additional equipment, including liquid handling robotics. The DMA technology can also be used in combination with direct dispensing of compounds, reagents, and even cells into individual droplets using an appropriate liquid dispenser. In this case, the volume of medium and number of cells in the droplets could be better controlled, which is beneficial for some applications. However, in order to be able to perform such dispensing in a fast and parallel manner compatible with high throughput, we would need new technical solutions.
We established a workflow for automated imaging of live cells directly in the droplets. Due to small volumes, suspension cells tend to locate close to the surface rather than being distributed throughout the whole droplet. We accounted for the small differences in cell localization by acquiring a Z stack of 10 slices and processing images using the “Extended Depth” algorithm. This gave us good-quality images for pattern recognition analysis. We also established the procedure for fixation of suspension cells in droplets, which can be followed by microscopic analysis directly on the DMA platform. Microscopy of fixed or live suspension cells is challenging, because cells are not fixed to the surface and constantly moving, making it difficult to acquire high-quality images. For this purpose, cells of a suspension nature have to be attached to the surface by using, for example, poly-
We developed an algorithm for automated counting of stained cells allowing for estimation of cell viability in a particular droplet. Estimation of viability using the microscopic approach might be more accurate than using bulk colorimetric or luminescence assays. Colorimetric methods such as MTT and MTS-based assays have been documented to exhibit cytotoxicity and give wrong viability estimation due to various nonspecific reactions.37,38 Luminescence ATP assays are more accurate but give information only about the amount of live cells without distinguishing between different cell death types. In contrast, microscopic analysis offers the possibility to extract multiple morphological features, giving more information about the drug effect on the cells.
To test the established workflow, we performed a dose-dependent study of cytotoxicity of antineoplastic drugs on Jurkat cells. The IC50 values of doxorubicin and daunorubicin known from the literature range from 0.004444 to 29.9 and from 0.02 to 1,39–42 respectively. Differences in IC50 values obtained on the DMA platform and those known from the literature are common and arise from differences in the experimental setup. The IC50 values obtained from different laboratories are known to vary and strongly depend on such parameters as platform, cell number, time of incubation, and readout assays.
43
We observed relatively high variability in cell response to drug concentrations from the middle of the effective dose range. It is accepted that the lower variance in the case of high or low doses of drug compared with middle doses can be observed.
44
Nevertheless, the Z′ factor calculated based on positive (a compound in concentration of 1000 μM) and negative (DMSO control) controls confirmed the suitability of the developed methodology for screening applications. We also analyzed the effect of vincristine, cytarabine, and etoposide on the viability of Jurkat cells (
Conclusions
Taken together, we have demonstrated the applicability of DMA system for miniaturized HTS of suspension cells. We utilized the DMA platform for culturing of suspension Jurkat cells and demonstrated homogeneous pipetting-free cell seeding and constant proliferation of cells in individual droplets of 3–80 nL volume from 2 to 5 days, depending on droplet size. We developed a simple methodology to perform parallel treatments, staining, and fixation of suspension cells in individual nanodroplets without losing the content of the droplets. A workflow for automated imaging of suspension cells in the droplets and developed algorithms for pattern recognition for image analysis were demonstrated. Finally, we tested the system by introducing Jurkat cells to antineoplastic drugs. We believe that the DMA screening platform carries great potential to be adopted for various miniaturized screening applications on cells of a suspension nature due to its unique properties allowing for single-step parallel manipulations with suspension cells, as well as automated fluorescent microscopy directly on the platform.
Footnotes
Acknowledgements
The authors are grateful to Dr. Christine Blattner (Institute of Toxicology and Genetics, KIT) for providing the Jurkat cell line.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The research was supported by the ERC starting grant (ID 337077-DropCellArray), ERC PoC grant (ID DLV-680913-CellScreenChip), and Helmholtz Association’s Initiative and Networking Fund (grant VH-NG-621).
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.