
Abstract
A wide linearity range analytical method for the determination of lenalidomide in patients with multiple myeloma for pharmacokinetic studies is required. Plasma samples were ultrasonicated for protein precipitation. A solid-phase extraction was performed. The eluted samples were evaporated to dryness under vacuum, and the solid obtained was diluted and injected into the high-performance liquid chromatography (HPLC) system. Separation of lenalidomide was performed on an Xterra RP C18 (250 mm length × 4.6 mm i.d., 5 µm) using a mobile phase consisting of phosphate buffer/acetonitrile (85:15, v/v, pH 3.2) at a flow rate of 0.5 mL · min−1. The samples were monitored at a wavelength of 311 nm. A linear relationship with good correlation coefficient (r = 0.997, n = 9) was found between the peak area and lenalidomide concentrations in the range of 100 to 950 ng · mL−1. The limits of detection and quantitation were 28 and 100 ng · mL−1, respectively. The intra- and interassay precisions were satisfactory, and the accuracy of the method was proved. In conclusion, the proposed method is suitable for the accurate quantification of lenalidomide in human plasma with a wide linear range, from 100 to 950 ng · mL−1. This is a valuable method for pharmacokinetic studies of lenalidomide in human subjects.
Keywords
Introduction
Multiple myeloma (MM) is a B-cell malignancy of plasma cells and represents the second most common hematological malignancy (about 10%). MM is characterized by the proliferation of plasma cells. Five-year relative survival rates have shown to diminish with increasing patient age; however, improved survival rates have been reported in newly diagnosed MM patients in recent years since the introduction of novel agents such as thalidomide, lenalidomide, and bortezomib.
Lenalidomide (LND; Revlimid; Celgene, Summit, NJ) is an immunomodulatory agent that has been shown to mediate direct toxicity and induce an immune response through marked activation of T and natural killer (NK) cells in MM patients. LND has antiangiogenic and antineoplastic properties, combined with a long-term immunostimulatory effect, which improve both cellular and humoral immune function. 1 LND has demonstrated significant clinical efficacy in the treatment of hematological cancers and is currently licensed in Europe and the United States, in combination with dexamethasone, for the treatment of patients with relapsed and refractory MM who have received at least one prior therapy.2,3 The recommended dose for MM treatment is 25 mg orally once daily, but it may be reduced if adverse events are observed.
High-performance liquid chromatography (HPLC) has been employed for the determination of LND in plasma. In two studies, a mass spectrometric detector has been used due to its high sensitivity and selectivity4,5; however this type of detector is not available in most laboratories because of its high cost. In the past years, fluorescence detection has been proposed as an alternative to mass detectors, but LND is not a fluorescent compound so precolumn derivatization with fluorescamine is necessary, which increases time of procedure and variability.6,7 On the other hand, none of the published methods has a linearity range that allows therapeutic drug monitoring of LND for pharmacokinetic studies. Accordingly, the development of a new alternative analytical method for the determination of LND in plasma with adequate sensitivity and selectivity, low cost, and wider linearity range is needed.
The purpose of this study was to develop and validate a wide linearity range, specific, accurate, precise, and sensitive analytical method for the determination of LND in plasma in patients with MM.
Materials and Methods
Reagents
All reagents were of analytical grade. LND, 3-(4-amino-1,3-dihydro-1-oxo-2H-isoindol-2-yl)-2,6-piperidinedione, was purchased from Cayman Chemical (Ann Arbor, MI) and stored at −20 °C. Sodium dihydrogen phosphate was purchased from Panreac (Barcelona, Spain). Purified water was purchased from Grifols (Barcelona, Spain). Ortophosporic acid was obtained from Fluka (St. Louis, MO). Acetonitrile and methanol were obtained from Scharlab (Barcelona, Spain). Human plasma samples were collected from healthy volunteers at Doctor Peset University Hospital (Valencia, Spain) and kept frozen at −20 °C until analysis.
Instrumentation and Chromatographic Conditions
An Agilent Technologies 1100 liquid chromatograph with a quaternary pump, a diode array detector, a thermostated column compartment, an autosampler, and a HP Compaq computer equipped with Agilent-Chemstation software was used (Agilent Technologies, Santa Clara, CA). The chromatographic separations were performed on an analytical column Xterra RP C18 (250 mm length × 4.6 mm i.d., 5-µm particle diameter) manufactured by Waters Corporation (Milford, MA). The column temperature was kept constant at 21 ± 2 °C. Separations were performed in isocratic mode. The mobile phase used for separation consisted of phosphate buffer/acetonitrile (85:15, v/v, pH 3.2) pumped at flow rate of 0.5 mL · min−1. The mobile phase was vacuum-filtered through 0.45-µm nylon membranes (Micron Separations, Westboro, MA) and degassed by ultrasonication prior to HPLC analysis. The samples (80 µL each) were injected through a Rheodyne valve (Rheodyne, Cotati, CA). The UV detector was set at 311 nm. The column was equilibrated for 30 min prior to injection of solutions. The peak area of LND was measured in each chromatogram. Retention time of LND was 10.8 min.
A pH meter (model 3510; Jenway, Staffordshire, UK) connected to a glass pH-electrode was employed. An analytical microbalance SC2 (Sartorius Mechatronics, S.A., Madrid, Spain) was used to weight LND, and an analytical balance (GF-200; A&D Instruments, Abingdon, UK) was used to weight other reagents.
Solution Preparation
Lenalidomide standard solution
An accurately weighed amount (0.5 mg) of LND was transferred into a calibrated volumetric flask, dissolved in acetonitrile, and completed to volume with the same solvent to produce a stock solution of 0.5 mg · mL−1. On the day of analysis, the stock solution was further diluted with water to obtain a working standard solution containing 2.0 µg · mL−1.
Phosphate buffer solution
A weighed amount of sodium dihydrogen phosphate (3.6 g) was dissolved in 1000 mL distilled water. The pH of the solution was adjusted to 3.2 ± 0.1 with orthophosphoric acid.
General Procedure
Accurately measured aliquots of LND working stock solution (2.0 µg · mL−1) were transferred into 10 separate assay tubes each containing 525 µL plasma. The volumes in all vials were adjusted, as necessary, to 1.0 mL with water. This resulted in a series of LND-spiked plasma solutions covering the working range of 100 to 950 ng · mL−1. A blank solution containing water and 525 µL plasma was also prepared.
The assay tubes were ultrasonicated for 20 min at room temperature and then centrifuged for 10 min at 3000 rpm.
A solid-phase extraction (SPE) was carried out before injecting the solutions in the chromatograph using cyano bonding cartridges (Discovery DSC-CN SPE Tube, bed weight 500 mg, volume 3 mL; Sigma-Aldrich, St. Louis, MO). The steps followed for the extraction of LND were (1) cartridge conditioning with methanol (2 mL), (2) solid-phase equilibration with 0.03 M phosphate buffer (pH 7.4) (2 mL), (3) load of serum or plasma spiked with LND (1 mL), (4) washout with 0.03 M phosphate buffer (pH 7.4) (2 mL), and (5) elution with mobile phase (2 mL).
The eluted samples were evaporated to dryness at 70 ± 1 °C under a vacuum of 600 mm Hg for 60 min in a Heidolph Synthesis 1 Multi-evaporator (Heidolph Instruments GmbH & Co.KG, Schwabach, Germany). The temperature used for the evaporation did not compromise the stability of the drug, according to several studies.8-10 The solid obtained was further diluted in 110 µL of mobile phase and centrifuged for 2 min at 10,200 rpm. The supernatant was transferred into an HPLC vial, and a volume of 80 µL of this solution was injected into the HPLC system. Peak area values of LND obtained at retention time of around 10.8 min were plotted versus the LND concentration. Blank samples treated in the same manner but using water instead of LND working stock solution were prepared.
Method Validation
Validation of the assay consisted of the assessment of linearity, lower limit of detection and quantitation (LOD and LOQ), specificity, intraday and interday accuracy and precision, recovery, robustness, and ruggedness of the method. The validation was performed following International Conference on Harmonization (ICH) guidelines. 11 Linearity was determined at 10 concentration levels of LND (100, 150, 250, 350, 450, 550, 650, 750, 850, and 950 ng · mL−1). The selected concentrations covered the range of expected plasma concentrations after a standard LND dose of 25 mg once daily.12-14 Correlation coefficient (r), y-intercept, and slope of regression line were estimated.
The specificity of the method was ascertained by evaluating the presence of interferences at the retention time of LND.
The within-run and between-run precision and accuracy were determined by analyzing quality control (QC) samples of four concentration levels covering the calibration range (100, 250, 450, and 950 ng · mL−1). Within-run accuracy and precision were determined by analyzing in a single run four QC samples per level. Between-run accuracy and precision were determined by analyzing QC samples in three runs on 3 different days. The mean estimated concentrations were compared with the nominal values of the QC samples, and the coefficient of variation (CV) and relative error (RE) were calculated to determine precision and accuracy, respectively. The mean back-calculated concentrations should be within 15% of the nominal values for the QC samples, except for LOQ, which should be within 20% of the nominal value. CV value should not exceed 15% for the QC samples, except for the LOQ, which should not exceed 20%. 8
There are three different methods for the determination of LOD and LOQ as indicated in ICH guidelines. However in this study, to determine the LOD and LOQ, the signal-to-noise ratio was calculated by comparing results for samples with known concentrations to blank samples. The LOQ is also defined as the lowest concentration that can be quantitated with acceptable precision and accuracy under the stated experimental condition.
The mean recoveries of LND were determined by comparison of the peak area of the plasma QC levels with standard solutions of equivalent concentrations in the mobile phase. An analysis of variance (ANOVA) was carried out to determine if significant differences exist between the recoveries of LND at different QC levels.
The robustness of the method was evaluated by making minor changes on the chromatographic parameters. To measure the extent, the most critical parameters were interchanged while keeping the other parameters unchanged. The chromatographic parameters were interchanged within the range of 1% to 10% of the optimum recommended conditions. The parameters involved were the pH of the phosphate buffer used in the mobile phase, the ratio between the components of the mobile phase, and column temperature.
The ruggedness of the method measures the influence of external factors, such as different instruments, analysts, and days, on results. It was evaluated by applying the recommended analytical procedures on the same HPLC system on different days on the analysis of a series of LND-spiked plasma samples.
Pharmacokinetic Study of Lenalidomide
Four blood samples were obtained from a patient with MM at 0.5, 1, 2, and 4 h after the administration of LND. A single 25-mg dose of LND was administered orally. Blood samples of 5 mL were collected at the selected times. The study had the approval of the Ethics Committee for Human Experimentation of Doctor Peset University Hospital of Valencia (Spain, code 14/012) and was performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki.
Results and Discussion
Linearity
Statistical analysis using least squares regression indicated excellent linearity for LND in the mentioned range (100–950 ng · mL−1). A good correlation between LND peak areas and drug concentration was observed with r2 ≥ 0.99 for all standard curves ( Table 1 ). To examine whether the intercept was significantly different from zero, the intercept was subjected to a t test. The value of t was obtained as 0.56 with a p value of 0.593. Thus, acceptance of the null hypothesis indicates that the intercept was not significantly different from zero. Therefore, there is no interference from the solvent used in the method. Mean slope and r2 were 0.425744 and 0.997, respectively. A representative chromatogram of an LND-spiked plasma sample of 100 ng · mL−1 (LOQ) is shown in Figure 1 . A representative chromatogram of a blank plasma sample is shown in Figure 2 .
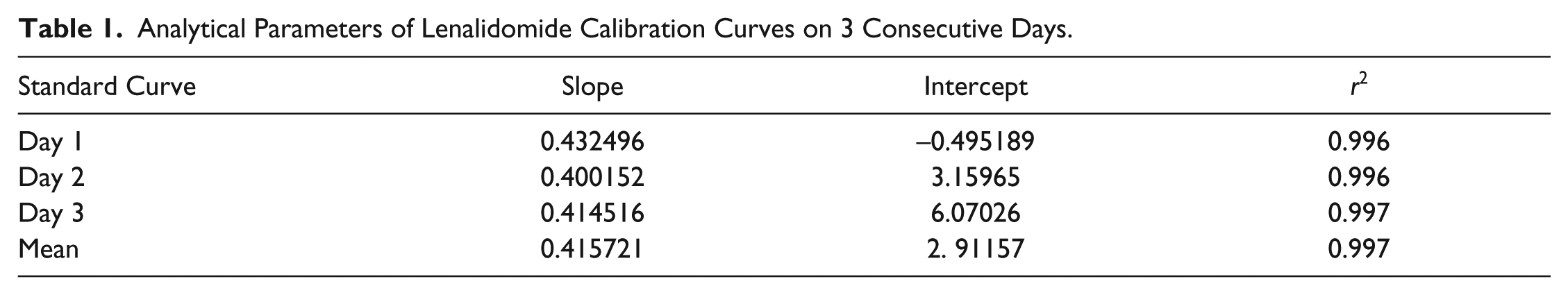
Analytical Parameters of Lenalidomide Calibration Curves on 3 Consecutive Days.
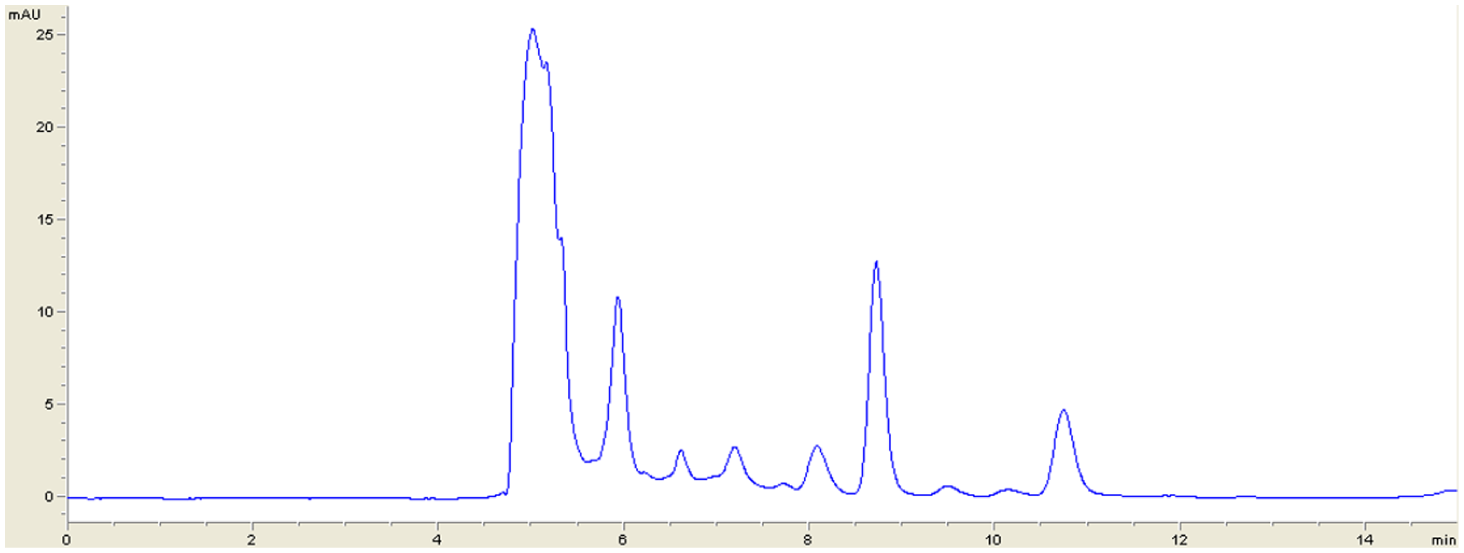
Representative chromatogram of a lenalidomide-spiked plasma sample of 100 ng · mL−1. The chromatogram corresponds to a sample of human plasma spiked with lenalidomide at a concentration of 100 ng · mL−1, which is the lower limit of quantitation of the developed method. The peak retention time of 10.8 min corresponds to lenalidomide.
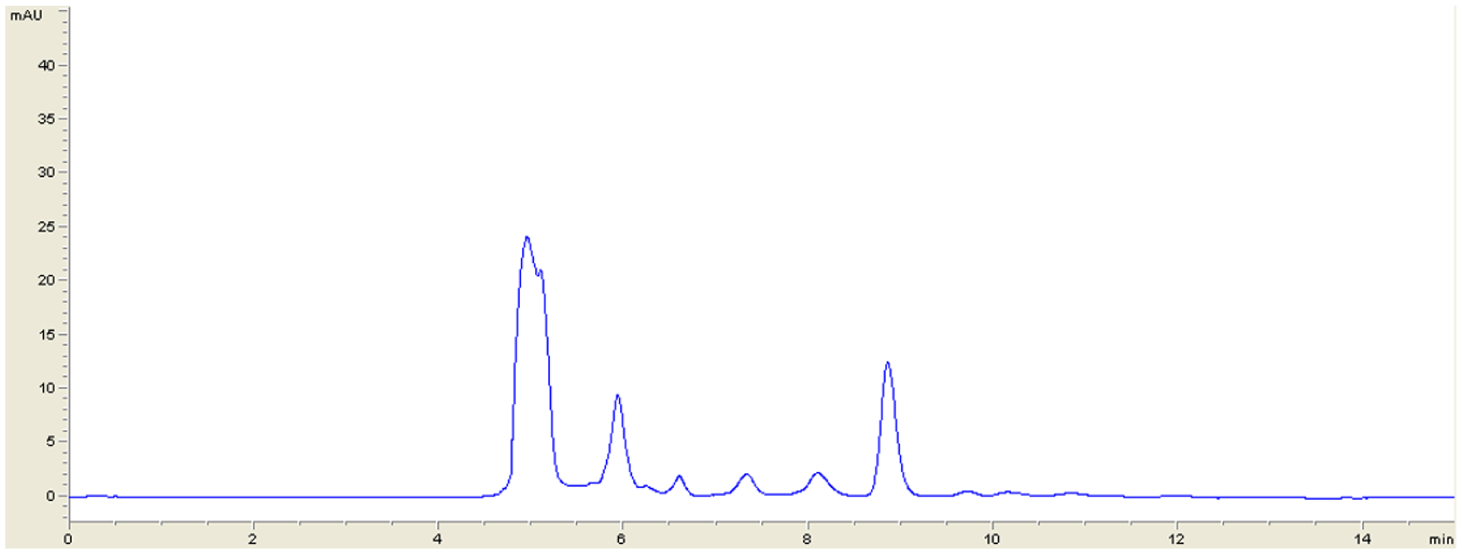
Representative chromatogram of a blank plasma sample. The chromatogram corresponds to a sample of human plasma from a healthy volunteer injected into the high-performance liquid chromatography system before spiking it with lenalidomide. No peak is observed at the lenalidomide retention time (10.8 min).
LOD and LOQ
The LOD and LOQ were calculated according to the ICH guidelines for validation of an analytical procedure based on the signal-to-noise ratio. Calculations on four replicate experimental injections showed that the LOD and LOQ were 28 and 100 ng · mL−1, respectively. The signal-to-noise ratio was near the expected value (LOD/LOQ = 28/100 ng · mL−1).
Accuracy and Precision
The within-run and between-run relative error (accuracy) and the CV (precision) are presented in Table 2 . The intraday accuracy and precision range were calculated to be −3.40% to 3.95% and 3.08% to 8.33%, respectively. The same parameters for interday evaluations were −1.79% to 7.59% and 0.88% to 7.43%, respectively. Accuracy and precision results were considered adequate.
Within-Run and Between Run Accuracy and Precision Results.
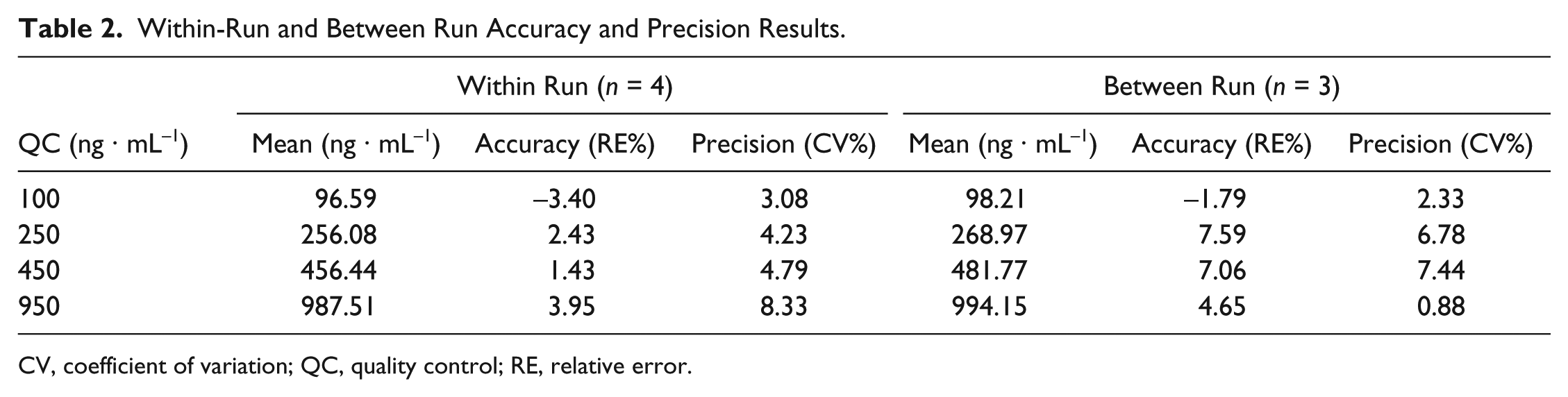
CV, coefficient of variation; QC, quality control; RE, relative error.
The recovery values were 79.4% to 92.4%, with a mean value of 85.98% ± 5.33%. The ANOVA results showed that there was no significant difference in the recoveries of LND at the four QC levels (p < 0.05), indicating that the described method is adequate for recovering LND.
Robustness and Ruggedness
The results revealed that the method was robust for these small changes in the parameters. However, increasing the pH value above 3.5 resulted in marked decrease in the detector signal.
As regards ruggedness, the variations in values of the capacity factor, retention time, and peak areas obtained each time were not significant.
Pharmacokinetic Study of Lenalidomide
A pharmacokinetic profile of LND obtained in a patient with MM after oral administration of a single dose of 25 mg is shown in Figure 3 . The concentrations obtained after oral administration were inside the range of concentrations covered by the calibration curve, and thus, the chromatographic method was considered adequate for its application in pharmacokinetic studies.
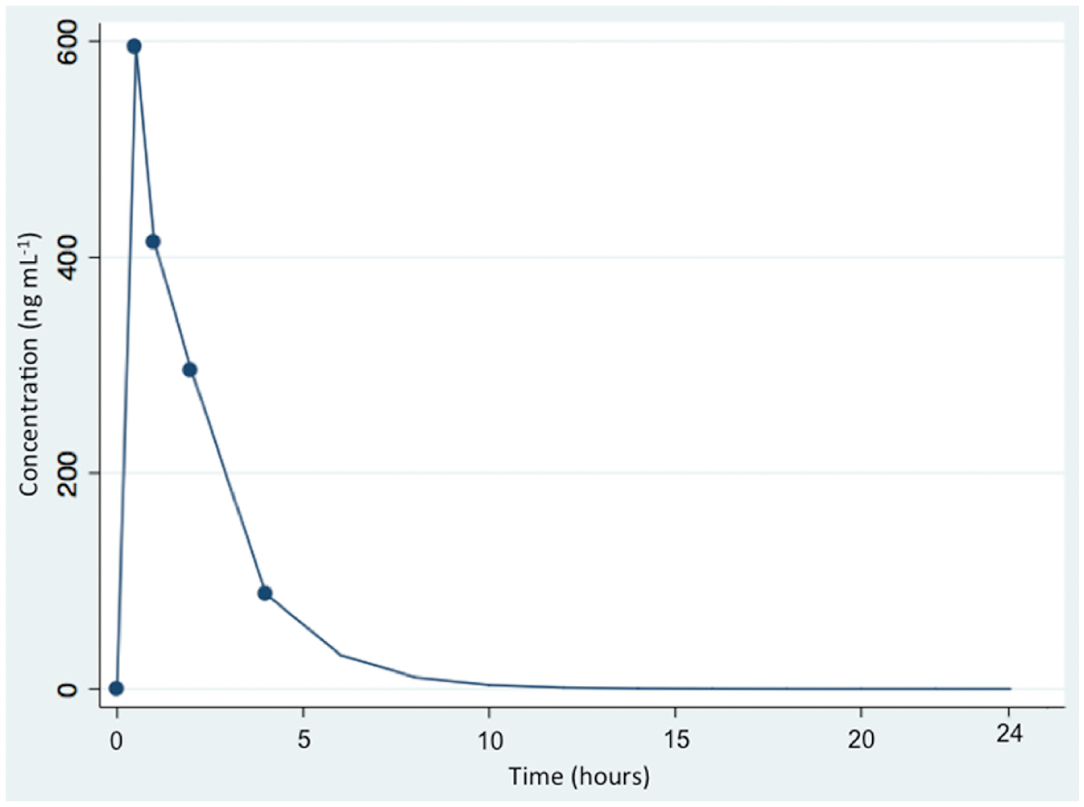
Pharmacokinetic profile of lenalidomide. The figure represents the pharmacokinetic profile of lenalidomide obtained in a patient with multiple myeloma after oral administration of a single dose of 25 mg lenalidomide.
In conclusion, an accurate and precise HPLC method with UV detection for determination of LND in human plasma has been successfully developed and validated. The chromatographic separation is based on a reversed-phase mechanism carried out under isocratic elution mode for only less than 15 min total runtime. The analytical results demonstrated that the proposed method is suitable for the accurate quantitation of LND in human plasma with a wide linear range, from 100 to 950 ng · mL−1. This is a valuable method for pharmacokinetic studies of LND in human subjects.
Footnotes
Acknowledgements
We thank the Hematology department for their collaboration.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: FISABIO foundation provided financial support (Project UGP-14-179).