
Abstract
Many biomarker-based diagnostic methods are inhibited by nontarget molecules in patient samples, necessitating biomarker extraction before detection. We have developed a simple device that purifies RNA, DNA, or protein biomarkers from complex biological samples without robotics or fluid pumping. The device design is based on functionalized magnetic beads, which capture biomarkers and remove background biomolecules by magnetically transferring the beads through processing solutions arrayed within small-diameter tubing. The process was automated by wrapping the tubing around a disc-like cassette and rotating it past a magnet using a programmable motor. This device recovered biomarkers at ~80% of the operator-dependent extraction method published previously. The device was validated by extracting biomarkers from a panel of surrogate patient samples containing clinically relevant concentrations of (1) influenza A RNA in nasal swabs, (2) Escherichia coli DNA in urine, (3) Mycobacterium tuberculosis DNA in sputum, and (4) Plasmodium falciparum protein and DNA in blood. The device successfully extracted each biomarker type from samples representing low levels of clinically relevant infectivity (i.e., 7.3 copies/µL of influenza A RNA, 405 copies/µL of E. coli DNA, 0.22 copies/µL of TB DNA, 167 copies/µL of malaria parasite DNA, and 2.7 pM of malaria parasite protein).
Keywords
Introduction
Biomarkers of infectious diseases are found at low concentrations compared to the abundance of human biomolecules and small molecules found in biological samples. Diagnostic techniques such as PCR and enzyme-linked immunosorbent assay (ELISA) are used to detect these biomarkers, but the presence of interfering molecules from the sample matrix or extremely low biomarker concentrations can negatively affect assay performance. To address these issues, extraction techniques are used to purify and concentrate the biomarker of interest prior to detection. In high-throughput central hospital diagnostic laboratories, biomarker extraction techniques have been highly automated for simultaneous processing of many patient samples. However, these large-scale instruments and techniques are impractical for use in small diagnostic laboratories, such as rural clinics. In these “low-sample-number” settings, an automated biomarker extraction device should be inexpensive, simple to use, capable of processing multiple samples at once, and flexible enough to allow multiple sample types and independent processing start times.
Both large- and small-scale biomarker extraction devices frequently use functionalized magnetic beads to isolate nucleic acids and proteins due to their customizable surface chemistry and simple, nonfluidic magnetic processing.1-5 These devices are typically well plate- or chip-based, and they use an external magnet to either keep the beads stationary within a plate or move the beads through an array of processing solutions. Plate-based devices require complex fluid pumping and movable robotics, and they are forced by design into batch-based processing. 1 In this device design, multiple samples are processed in parallel on a plate, and an entire plate is usually filled before processing. These systems work well in high-throughput laboratories, but they are inefficient in low-sample-number settings. Magnetic beads have been used in chip devices to eliminate the need for fluid pumping, but the format requires multiple solution outlets and precise fluid manipulations. 5 Furthermore, these devices do not have modular components for extraction of multiple biomarker classes.
In this report, we describe a device that can be used to extract and concentrate RNA, DNA, or protein biomarkers from complex samples in an asynchronous, random-access format. In a low-sample-number setting, this device significantly improves throughput because each sample is processed independently of other sample types and multiple samples can be processed in parallel. The device ( Fig. 1 ) consists of a circular plastic cartridge that rests on both a roller controlled by a programmable motor and a support bar containing six neodymium magnets. To perform an extraction, the sample is incubated with magnetic beads to capture the biomarker of interest, and then it is loaded into a length of small-diameter plastic tubing containing preloaded processing solutions specific to the RNA, DNA, or protein biomarkers. This tubing is wrapped around the device cartridge so that as the cartridge rotates, the biomarker-bound magnetic beads are transferred through the processing solutions by the stationary external magnet. Biomarker extraction in this format is enabled by our previously described surface tension valves, or immiscible fluid separators, which are used to sequentially array processing solutions within a single length of small-diameter tubing.6-9 This simple format has advantages over other biomarker extraction procedures because multiple biomarker classes can be extracted on the same device by simply using different bead surface chemistries and processing solutions. Furthermore, because of the continuous cartridge roller design, cartridges can be added to or removed from the device asynchronously and without stopping the device. This random-access operation increases throughput without the need to wait for an adequate number of samples for batching biomarker extractions. The device requires little power and is simple to operate. In this report, we validated the performance of this device for the extraction of RNA, DNA, and protein using a panel of surrogate patient samples prepared by the Program for Appropriate Technology in Health (PATH).
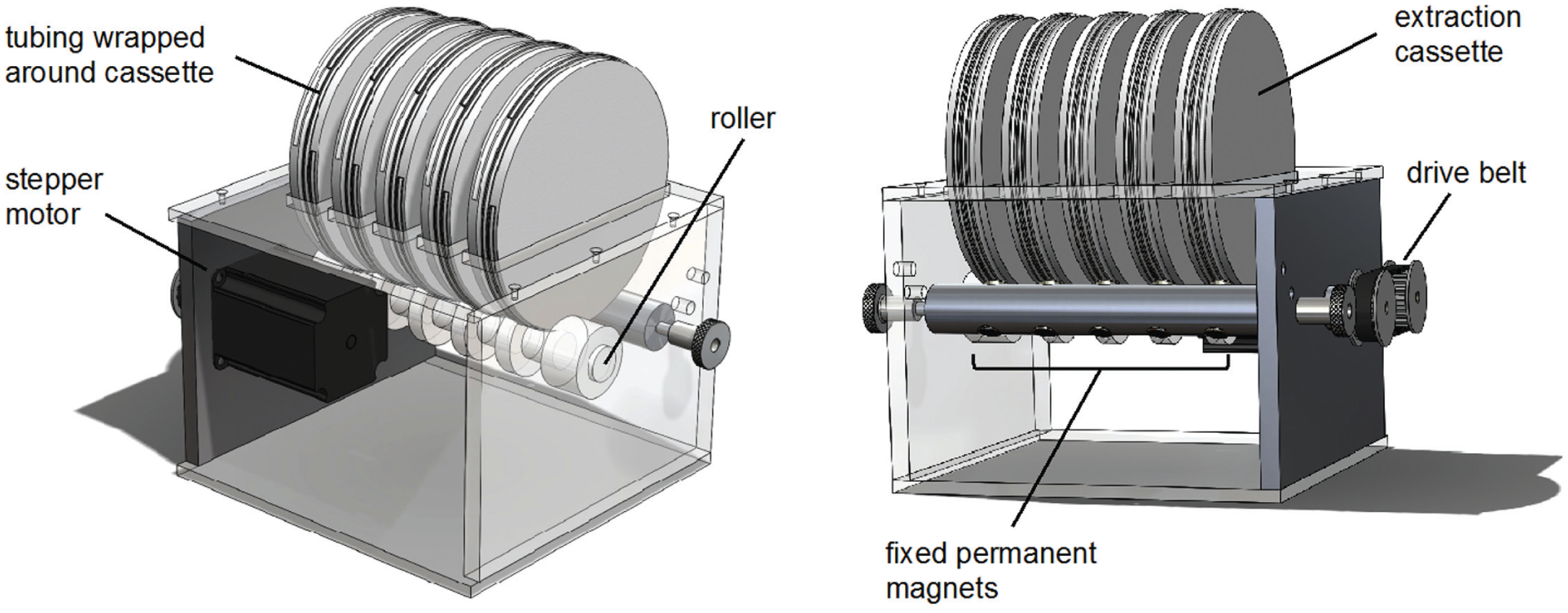
The automated biomarker extraction device. The tubing is wrapped around the outer circumference of extraction cassettes. A stepper motor rotates the rollers, which rotate the extraction cassettes past fixed permanent magnets. The stepper motor drive controller, Arduino Rev3 programmable controller, and power supply are not shown.
Materials and Methods
Materials for Device Fabrication
The Arduino UNO Rev3 programmable controller was purchased from Arduino (#A000066). The stepper motor drive controller (#STR4), stepper motor (#HT23-401), and power supply (#PS150A24) were purchased from Applied Motion Products (Watsonville, CA). Cylindrical magnets were purchased from SuperMagnetMan (#Cyl0360 N40; Birmingham, AL). The components were assembled using aluminum components made in-house by the Vanderbilt Physics and Astronomy Machine Shop. The plastic cassettes were designed and printed in-house using SolidWorks software and a NovaCopy ProJet HD 3000 Plus 3D printer (NovaCopy, Nashville, TN). The STL file for the plastic cassette is available as Supplemental Materials online.
Design and Operation of the Automated Biomarker Extraction Device
The automated biomarker extraction device was designed to transport magnetic beads through a series of processing solutions contained within 1.6 mm inner-diameter (i.d.) tubing by rotating an extraction cassette (i.e., a plastic disc with extraction tubing wrapped around the circumference) past a fixed magnet using a simple stepper motor ( Fig. 1 ). The cassette is a plastic disc that is 14 cm in diameter and 1.9 cm thick, with a continuous channel on the outer edge to secure the assay tubing around the disc circumference. In this design, individual cassettes are placed onto a rotating shaft to drive the rotation of the cassettes past a second shaft containing fixed magnets. As the disc rotates, the magnetic beads within the assay tubing are held in place by the magnets while the processing solutions contained in the assay tubing move past the beads. An open-top roller-drive design was chosen over an axle-drive design to facilitate asynchronous, random-access loading and processing of independent cassettes on the device.
The Arduino UNO Rev3 programmable controller was used to send logical input commands to the motor controller to regulate the direction, speed, and timing of the drive motor. The Arduino code was developed to allow the operator to input desired rotational distance and velocity. The default commands used to operate the Arduino UNO controller and a device schematic are provided as Supplemental Materials. Two timing gears and a ribbed belt attach the motor to a roller on which the cassettes sit. As the roller rotates, friction between the roller and the cassettes causes the cassettes to rotate past the panel of fixed magnets. The rotation processes magnetic beads through the array of solutions within the tubing.
Materials for Biomarker Extraction and Detection
Sample panels infected with influenza A, Esherichia coli (E. coli), Mycobacterium tuberculosis (TB), and Plasmodium falciparum (malaria parasite) were prepared and provided by PATH. Primers, probes, and reference standard templates were purchased from Integrated DNA Technologies (Coralville, IA). SuperScript III RT/Platinum TAQ mix (#11732-088) and Dynabeads MyOne Silane magnetic beads (#37002D) were purchased from Life Technologies (Carlsbad, CA). Quanta PerfectA 2× mastermix was purchased from VWR (#84010; Quanta Biosciences, Gaithersburg MD). Lysozyme from chicken egg white was purchased from Sigma-Aldrich (#6876; St. Louis, MO). Neodymium magnets were purchased from Emovendo (Petersburg, WV). Filtration swabs (#5001.02) were purchased from Salimetrics (State College, PA). Ni-NTA Magnetic Agarose Beads (#36111) were purchased from Qiagen (Venlo, the Netherlands). Antibodies for the pfHRPII ELISA (#ab9206 and ab30384) were purchased from Abcam (Cambridge, UK). 3,3′,5,5′-tetramethylbenzidine (TMB) One solution (#G7431) was purchased from Promega (Fitchburg, WI). Recombinant HRPII (#AGPF-55) was purchased from Immunology Consultants Laboratory (Portland, OR). Fluorinated ethylene propylene (FEP) tubing was purchased from McMaster Carr (#2129T11 and 9369T46; Elmhurst, IL). Tygon R-3603 tubing (#14-169-1B) and Hemato-Seal tube-sealing compound (#02-678) were purchased from Fisher Scientific (Hampton, NH). All other materials and reagents were purchased from Sigma-Aldrich or Fisher Scientific.
Development of the Automated Extraction Cassette Processor
The initial automated extraction process was developed using samples from our recent work with detecting TB DNA in human urine samples. 7 For these studies, a 30 cm length of 1.6 mm i.d. FEP tubing was preloaded with processing solutions according to Bordelon et al. 7 Briefly, the tubing was loaded by pipetting 50 µL nuclease-free water, 300 µL 70% ethanol, and 300 µL DNA precipitation buffer (80% ethanol, 5 mM potassium phosphate, pH 8.0) into one end. Each solution was separated from the next by a 6 mm air valve. A 500 µL human urine sample spiked with 5 × 108 copies of a 140 nucleotide TB IS6110 DNA sequence was combined with 500 µL DNA–silica adsorption buffer (4 M guanidine thiocyanate, 25 mM sodium citrate, pH 7.0) and 6 × 108 silica-coated magnetic beads (20 µL suspension), and placed on a laboratory rotisserie for 5 min. Following nucleic acid adsorption, the tube contents were pipetted into the end of the prepared small-diameter tubing, and both ends were sealed with tube-sealing compound. The prepared tubing was processed one of three ways: manually as described by Bordelon et al. 7 , using a continuous single-speed automated approach, or using a continuous multispeed automated approach.
For automated processing, the prepared tubing was loaded onto the extraction cassette and placed on the device. During single-speed processing, the extraction device was programmed to rotate the cassette at a single fixed speed to continually pull the beads from one end of the tubing to the other with no mixing. The bead procession rate was set to 5°/s, 10°/s, 15°/s, or 20°/s (~0.6 cm/s, ~1.2 cm/s, ~1.8 cm/s, and ~2.4 cm/s, respectively), and the extraction was allowed to proceed until the magnetic beads exited the final chamber. The tubing was removed from the cassette, and the elution chamber was excised for downstream quantification by PCR using a Rotor-Gene Q thermal cycler.
During multispeed processing, the extraction device was programmed to rotate the cassette at two alternating speeds and distances. The first phase was short and slow, designed to transport the beads down the length of the tubing, and the second phase was long and fast to mix the beads within each processing solution by disassociating them from the magnetic field. During bead transfer phases, the motor rotated the cassette 10° (~1.2 cm) at a relatively slow rate [i.e., 5°/s (~0.6 cm/s) for influenza RNA, E. coli DNA, and TB DNA; 4°/s (~0.5 cm/s) for malaria parasite DNA; and 3°/s (~0.4 cm/s) for malaria parasite protein]. Based on results from the single-speed experiments described above, the slow step of 5°/s was chosen as a starting point for the multispeed method. Because magnetic beads of lower magnetic susceptibility were used for the malaria culture samples, slightly slower speeds were chosen for the malaria culture samples to ensure that all magnetic beads were effectively removed from the complex sample solution. During the bead-mixing phase, the cassette rotated 715° (just short of two complete rotations) at a relatively fast rate [i.e., 150°/s (~18.3 cm/s) for influenza RNA, E. coli DNA, and TB DNA; 80°/s (~9.8 cm/s) for malaria parasite DNA; and 60°/s (~7.3 cm/s) for malaria parasite protein]. The fast rotation was followed by a 5 s pause to collect the beads before the bead transfer phase was repeated. The extraction was allowed to proceed until the magnetic beads were deposited into the final chamber. After extraction, the tubing was removed from the cassette, and the eluate was quantified by reverse transcriptase quantitative PCR (RT-qPCR), qPCR, or ELISA, as described below.
Nucleic Acid Quantification by qPCR and RT-PCR
Nucleic acids extracted from the PATH panel samples were quantified by RT-qPCR or qPCR as appropriate according to protocols adapted by PATH from existing studies.10-13 The primer–probe sequences and cycling conditions for each nucleic acid biomarker are provided in Table 1 . Influenza A RNA was amplified via RT-qPCR using the SuperScript III RT/Platinum Taq mix in 25 µL reaction volumes with a final primer concentration of 800 nM and a final probe concentration of 150 nM. DNA from E. coli, TB, and malaria parasite was amplified via PCR using Quanta PerfectA 2× mastermix in 25 µL reaction volumes. Final primer concentrations were 800 nM, 300 nM, and 800 nM for E. coli, TB, and malaria parasite, respectively. The final probe concentration was 150 nM for all reactions. For quantification, each reaction was performed in parallel with a standard curve for each biomarker target. Standard curves were prepared by serially diluting synthetic DNA amplicons in Tris–EDTA buffer. The sequence of each amplified gene was purchased from Integrated DNA Technologies. Tenfold dilutions were prepared from 107 copies/µL to 10 copies/µL. No-template controls were also performed, with no DNA added. Thermal cycling was performed using the Qiagen Rotor-Gene Q or Roche LightCycler 96, and cycle threshold (Ct) values were calculated using the provided software.
The Primer and Probe Sequences and Thermal Cycling Conditions Used for the Quantification of Nucleic Acids Extracted from Validated Panel Samples.
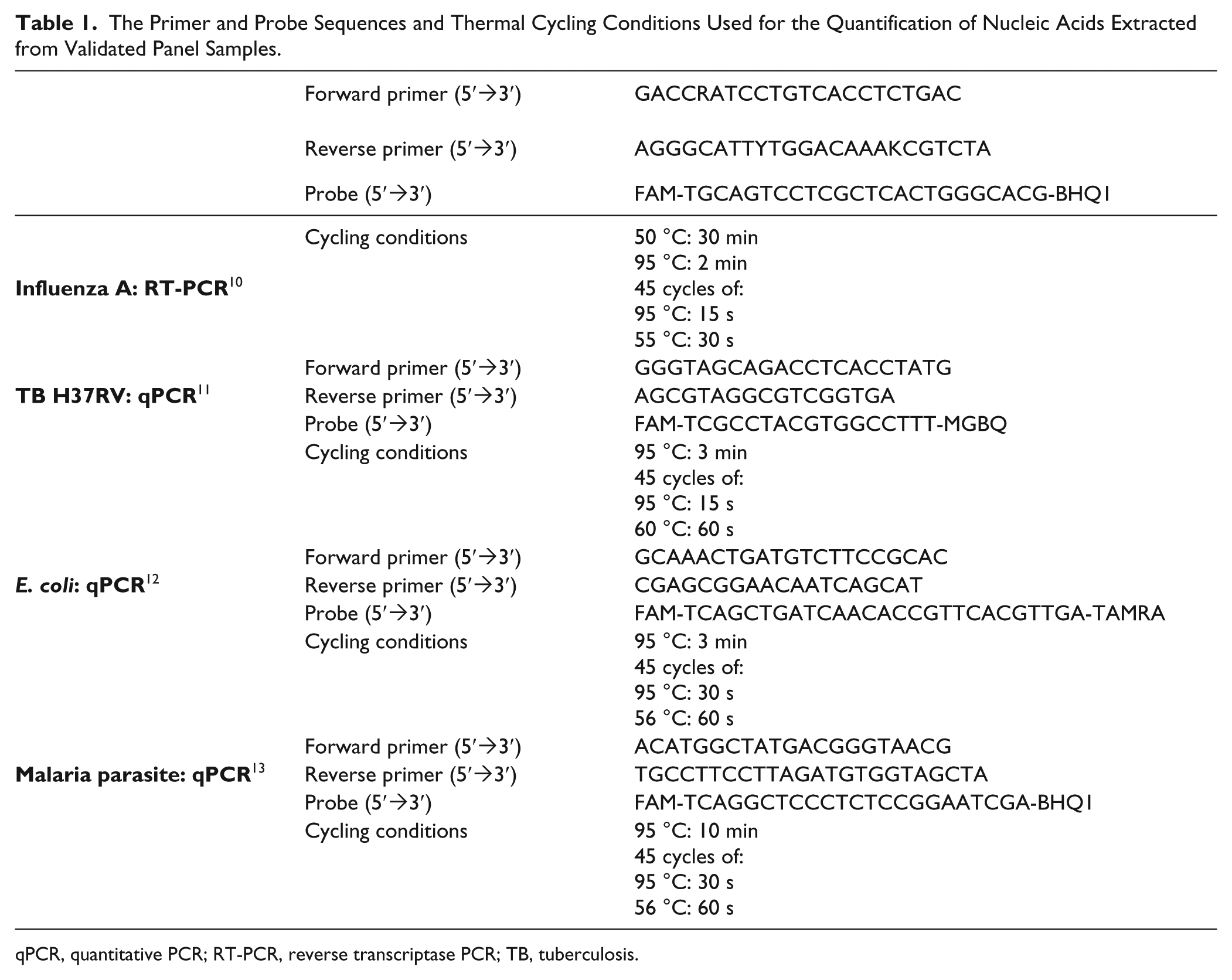
qPCR, quantitative PCR; RT-PCR, reverse transcriptase PCR; TB, tuberculosis.
Protein Quantification by ELISA
Extracted pfHRPII protein was quantified by ELISA in 96-well Immulon 2 HB plates according to Davis et al. 9 Once the plate was developed, absorbance was measured at 450 nm using a BioTek Synergy H4 microplate reader, and the protein was quantified by referencing a standard curve made using recombinant HRPII (rcHRPII) purchased from Immunology Consultants Laboratory (Portland, OR).
Validation Using Surrogate Patient Sample Panels from PATH
The efficacy of the automated biomarker extraction device was tested using five panels of surrogate patient samples prepared and validated by PATH. Each panel contained surrogate patient samples spiked with concentrations of pathogen corresponding to a biologically relevant low, medium, or high level of infection. Negative control samples containing no pathogen were also provided for each sample type. The sample panels tested are listed in Table 2 . Each sample panel was received frozen on dry ice and stored at −80 °C until used.
Summary of Validated PATH Surrogate Sample Panels Tested in This Study.
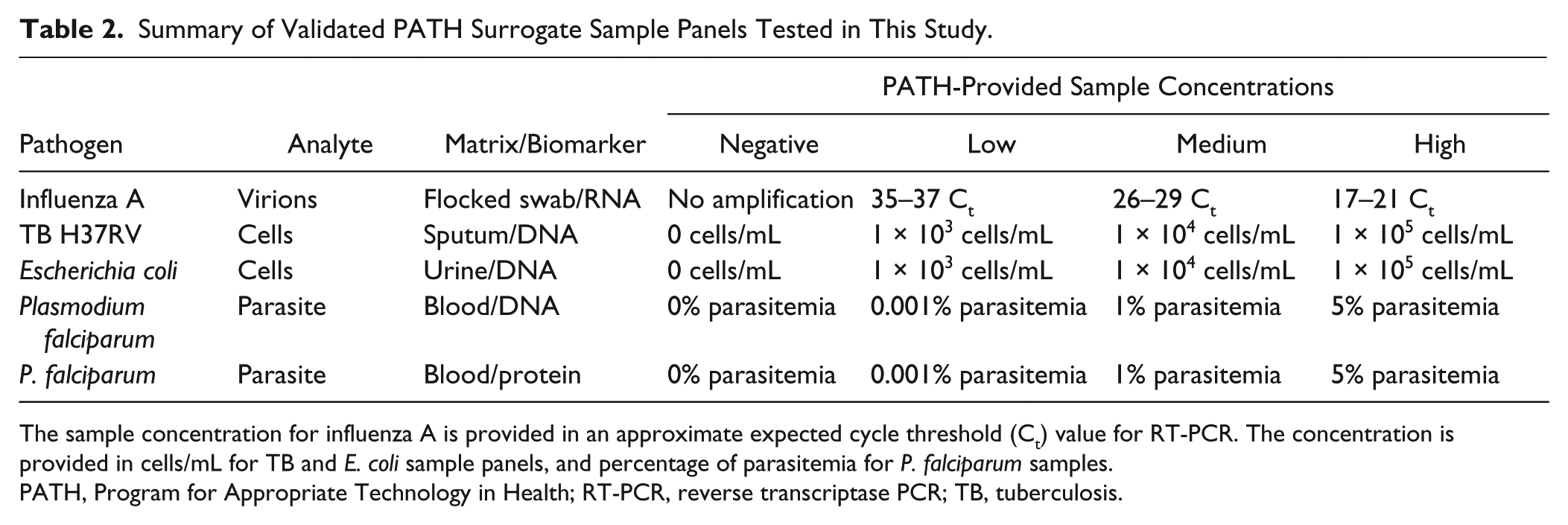
The sample concentration for influenza A is provided in an approximate expected cycle threshold (Ct) value for RT-PCR. The concentration is provided in cells/mL for TB and E. coli sample panels, and percentage of parasitemia for P. falciparum samples.
PATH, Program for Appropriate Technology in Health; RT-PCR, reverse transcriptase PCR; TB, tuberculosis.
Extraction of Influenza A RNA from Flocked Swabs
Assay tubing was prepared in a 15 cm length of 1.6 mm i.d. FEP tubing. The tubing was loaded by pipetting 50 µL nuclease-free water, 300 µL 70% ethanol, and 300 µL RNA precipitation buffer (80% ethanol, 5 mM potassium phosphate, pH 8.0) into one end. Each solution was separated from the next with a ~6 mm gap of air.
Influenza A virions were eluted from the swab by soaking in 180 µL of sterile phosphate buffered saline for 15 min in a 2 mL microcentrifuge tube. The flocked swab was discarded, and the contents of the tube were vortexed briefly. The sample was combined with 300 µL RNA–silica adsorption buffer (5.7 M guanidine thiocyanate, 25 mM sodium citrate, 1% 2-mercaptoethanol, pH 7.0) and 6 × 108 silica-coated magnetic beads (20 µL suspension), and vortexed thoroughly. After the addition of 300 µL of ethanol, the tube was placed on a laboratory rotisserie for 5 min. Following nucleic acid adsorption, the tube contents were pipetted into the end of the preloaded tubing, both ends were sealed with tube-sealing compound, the tubing was loaded on the extraction cassette, and extraction was carried out as described above. After about 5 min of automated extraction, the tubing was removed from the cassette, and the RNA in the final solution was quantified by RT-PCR as described above.
Extraction of E. coli DNA from Human Urine
Assay tubing was prepared in a 10 cm length of 1.6 mm i.d. FEP tubing. The tubing was loaded by pipetting 50 µL nuclease-free water, 300 µL 70% ethanol, and 300 µL DNA precipitation buffer (80% ethanol, 5 mM potassium phosphate, pH 8.0) into one end. Each solution was separated from the next by a ~6 mm air valve.
A 1 mL E. coli–spiked urine sample was centrifuged for 5 min at 10,000 × g in a 1.5 mL microcentrifuge tube, and the supernatant was discarded. The E. coli cells were lysed by adding 100 µL lysis buffer (50 mM Tris-HCl, 10 mM EDTA, 5% Triton X-100, 100 mM NaCl) and 25 µL lysozyme solution (10 mg/mL) at 25 °C. The tube was vortexed briefly and placed on a laboratory rotisserie for 20 min. The lysed cells were centrifuged for 5 min at 10,000 × g to remove cell debris, and the supernatant was retained for nucleic acid extraction. The sample was combined with 300 µL DNA–silica adsorption buffer (4 M guanidine thiocyanate, 25 mM sodium citrate, pH 7.0) and 6 × 108 silica-coated magnetic beads (20 µL suspension), and placed on a laboratory rotisserie for 5 min. Following nucleic acid adsorption, the tube contents were pipetted into the end of the preloaded tubing, both ends were sealed with tube-sealing compound, the tubing was loaded on the extraction cassette, and extraction was carried out as described above. After 5 min of automated extraction, the tubing was removed from the cassette, and the DNA in the final solution was quantified by PCR as described above.
Extraction of Mycobacterium tuberculosis DNA from Synthetic Sputum
Assay tubing was prepared in a 70 cm length of 1.6 mm i.d. FEP tubing. The tubing was loaded by pipetting 50 µL nuclease-free water, 300 µL 70% ethanol, 300 µL RNA precipitation buffer (80% ethanol, 5 mM potassium phosphate, pH 8.0), and 300 µL chaotropic wash buffer (4 M guanidine hydrochloride, 25 mM sodium citrate, pH 7.0) into one end. Each solution was separated from the next by a ~6 mm air valve.
A 500 µL TB-infected synthetic sputum sample was combined with TB lysis/binding buffer (4 M GuSCN, 25 mM sodium citrate, 4.9% Triton X-100, 0.2% sodium dodecyl sulfate, pH 7.0) and 6 × 108 silica-coated magnetic beads (20 µL suspension) in a 2 mL microcentrifuge tube. The tube was vortexed for 10 min on a Fisher Vortex Genie 2 set to speed 4 to lyse the bacterial cells and capture released nucleic acids. This lysis method was developed by combining elements from multiple methods described for lysis of mycobacteria.14-16 Following nucleic acid adsorption, the tube contents were pipetted into the end of the preloaded tubing, both ends were sealed with tube-sealing compound, the tubing was loaded on the extraction cassette, and extraction was carried out as described above. After 5 min of automated extraction, the tubing was removed from the cassette, and the DNA in the final solution was quantified by PCR as described above.
Extraction of Plasmodium falciparum DNA from Human Blood Culture
Assay tubing was prepared in a 43 cm length of 2.36 mm i.d. FEP tubing. The tubing was loaded by pipetting 50 µL elution buffer (10 mM Tris pH 8.0, 1 mM EDTA, 0.05% Tween-20), 300 µL 70% ethanol, and 300 µL chaotropic wash buffer (80% ethanol, 640 mM guanidine thiocyanate, 1.6 mM Tris pH 8.0, 160 µM EDTA, 0.08% Triton X-100) into one end. Each solution was separated from the next by a ~6 mm air valve.
A 100 µL sample of malaria parasite-infected human blood culture (5% hematocrit) was combined with 300 µL lysis buffer (4 M guanidine thiocyanate, 10 mM Tris HCl pH 8.0, 1 mM EDTA, 0.5% Triton X-100) and incubated at room temperature for 10 min with occasional vortexing to lyse red blood cells. The lysed samples were then filtered using one-fourth of a Salimetrics swab, and the sample was expressed from the swab by using a 5 mL plastic syringe. After the addition of 100 µL of 100% isopropanol and 7.5 × 108 silica-coated magnetic beads (25 µL suspension) to the filtered sample, it was incubated for 3 min at room temperature. Following nucleic acid adsorption, the tube contents were pipetted into the end of the preloaded tubing, both ends were sealed with tube-sealing compound, and the assay was completed using both a manual magnetic bead–based extraction method and the two-speed automated extraction method.
For manual processing, the beads were collected within the sample chamber using a 1-inch neodymium magnet. The beads were slowly pulled across the air valve into the first wash chamber. The magnet was moved back and forth quickly along the wall of the tubing to mix the beads within the first wash chamber for 30 s. The beads were then pulled into the second wash chamber and mixed the same way for 30 s. Finally, the beads were pulled into the elution chamber, where they were mixed for 3 min using the magnet. The beads were then removed from the elution chamber, and the DNA in the final solution was quantified by qPCR as described above.
For automated processing, the tubing was loaded on the extraction cassette, and extraction was carried out as described above. After 5 min of automated extraction, the tubing was removed from the cassette, and the DNA in the final solution was quantified by qPCR as described above.
Extraction of Plasmodium falciparum HRPII from Human Blood Culture
Assay tubing was prepared in a 24 cm length of 1.6 mm i.d. Tygon R-3603 tubing. The rounded end of a 200 µL PCR tube was cut off, and the top of the cap was punctured with a 27½ gauge needle to serve as an air release valve. The modified tube was then inserted into one end of the Tygon tubing. Ten microliters of elution buffer (50 mM potassium phosphate, pH 8.0, 300 mM NaCl, 1 M imidazole, 0.05% Tween-20) was pipetted into the opposite end of the tubing, which was then sealed with tube-sealing compound. Mineral oil valves (25 µL mineral oil) were added alternately with three 100 µL wash chambers (50 mM phosphate buffer, pH 8.0, 300 mM NaCl, 125 mM imidazole, 0.05% Tween-20) down the length of the tubing using a 27½ gauge syringe. Finally, mineral oil was added between the final wash chamber and inserted PCR tube to seal off the tubing from air.
A 100 µL sample of infected blood culture (5% hematocrit) was lysed with 100 µL of lysis buffer (100 mM potassium phosphate pH 8.0, 600 mM NaCl, 250 mM imidazole, 2% Triton X-100) in a 1.5 mL microcentrifuge tube. Following lysis, the sample was filtered through a 1 mL syringe fitted with glass wool to removed lysed cell debris. The filtered sample was mixed with 1.5 × 103 magnetic Ni(II)NTA agarose beads (10 µL suspension) in the modified PCR tube, which was placed on a laboratory rotisserie for 10 min to ensure mixing of the beads throughout the sample. After the incubation period, the PCR tube was reinserted into the extraction tubing and the beads were pulled into the adjacent mineral oil valve using a donut-shaped neodymium magnet. Finally, the PCR tube was removed, and the extraction tubing was sealed with tube-sealing compound. The tube contents were pipetted into the end of the preloaded tubing, both ends were sealed with tube-sealing compound, the tubing was loaded on the extraction cassette, and extraction was carried out as described above. After 5 min of automated extraction, the tubing was removed from the cassette, and the protein in the final solution was quantified by ELISA as described above.
Results and Discussion
In this report, we describe the development and evaluation of an automated device that extracts nucleic acid and protein biomarkers from surrogate patient samples. The device was designed to move biomarker-bound magnetic beads through a series of processing solutions separated by surface tension valves within small-diameter plastic tubing by rotating the tubing past a fixed permanent magnet ( Fig. 1 ). This device features fewer user steps, increased throughput, and enhanced reproducibility compared to the manual (i.e., operator-dependent) extraction method we have reported previously.6,7,9
Reproducing the magnetic bead processing and mixing steps of the manual extraction represented one of the significant challenges of the automated system. Initial device development and optimization were performed using samples of TB DNA in urine. Recent findings described in the literature indicate that the detection of TB DNA in urine may hold promise as a simple means for identifying a TB infection compared to TB biomarkers in sputum.17-19 Furthermore, this sample matrix was used in our recent work to develop a manual extraction method and provided a good performance comparison. 7 Motor drive programs of increasing complexity were evaluated to determine the effects of automated processing on biomarker recovery. The first motor-driven program was a simple procession of the beads from one end of the tubing to another without their dispersion. Using this approach, recovery of a spiked DNA biomarker was inversely proportional to the speed at which the beads were processed through the cassette tubing ( Fig. 2A ). The maximum biomarker recovery was 2 ± 0.09 × 107 DNA copies when the beads were pulled through the processing solutions at the slowest processing speed of 5 degrees/s (~0.6 cm/s), but this recovery was about sixfold lower than for a manual extraction, which recovered 1.2 ± 0.5 × 108 copies ( Fig. 2B ). These results indicate that the single-speed extraction process is relatively ineffective, likely because of the lack of bead dispersion in each processing solution.
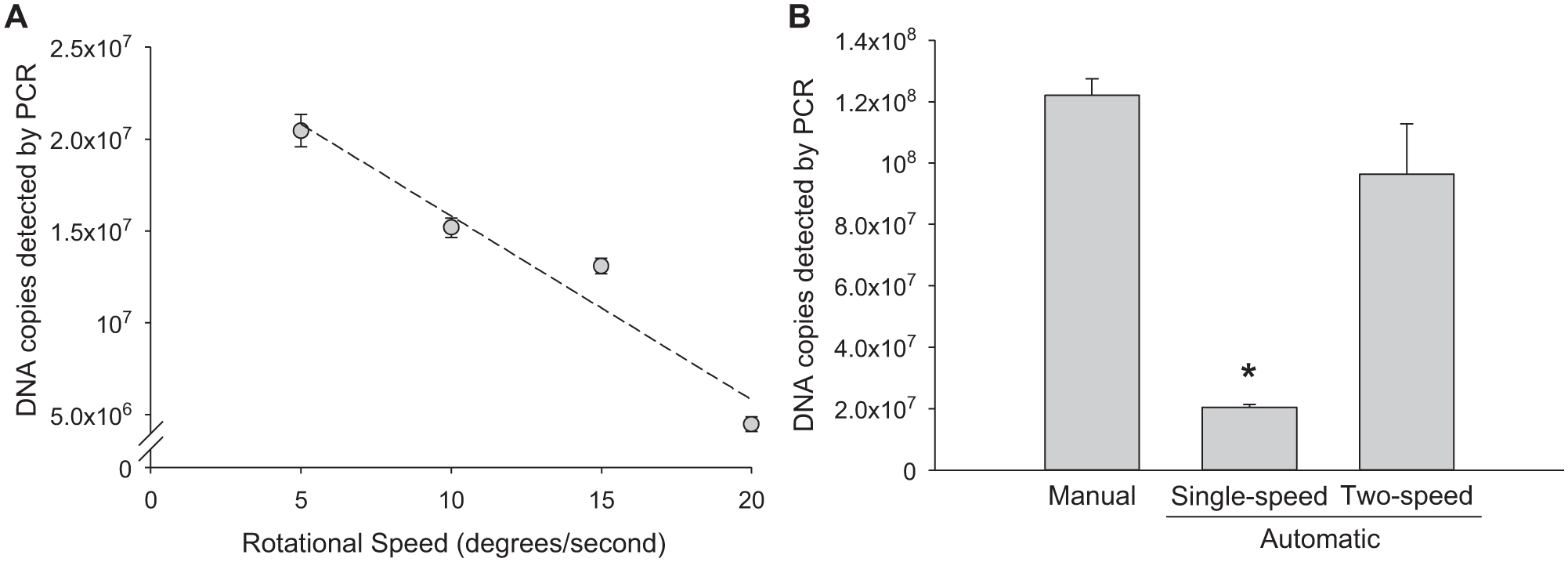
(
To replicate the bead dispersion of the manual extraction process, a more complex program was needed. One method that was considered was a program that slowly moved magnetic beads into processing solutions, then repeatedly reversed the motor direction to disperse the beads throughout the solution. The challenge with this method was that the motor would have to be programmed to process the beads based on the precise positions of the solutions and valves, so that beads would only be dispersed within the wash solutions. This would require a method of active feedback of solution and valve positions or highly reproducible positioning of solutions and valves within the extraction tubing. Furthermore, this position-dependent method would require different processing steps for each biomarker class and would not be tolerant of variations in the volumes of each sample or wash solution volume.
To overcome the challenges associated with a position-dependent extraction program, a position-agnostic program based on single-direction, two-speed processing was developed and evaluated. In this approach, the motor turns the assay tubing slowly past the magnet to move the beads forward ~10° (~1.2 cm), and then spins rapidly in the same direction for nearly two complete turns (715°). During the rapid spin, the beads become disassociated from the magnet and are dispersed throughout the processing solution. The beads are then collected with a short pause step, followed by an additional slow movement forward another ~10° (~1.2 cm). The process repeats until the beads reach the elution chamber. Using this approach, DNA biomarker recovery was 9 ± 2 × 107 copies, which was comparable to that of the manual extraction process ( Fig. 2B ). Variations in tubing contents, sample and wash buffer volumes, and surface tension valve lengths had no effect on biomarker recovery using this position-agnostic, two-speed processing method. Because the rotation of the alternating speeds is always in the same direction, the program operates independently of extraction cassette alignment. One advantage of this design over a traditional batched format is that multiple extraction cassettes can be loaded onto and removed from the device independently and asynchronously, thus decreasing the time-to-result for individual assays and increasing throughput. Furthermore, the cassettes can be processed without direct user attention, because the beads inevitably reach the sealed end of the tube and remain in the final solution until the individual cassette is removed from the device.
The automated device was compared to our previously developed manual extraction method7,9 using malaria DNA in blood culture. Nine samples from each panel concentration were extracted using both the manual method and the two-speed automated method. As shown in Figure 3 , the amount of DNA extracted using each method correlated with the malaria pathogen concentrations and extracted enough DNA to be detected at the lowest pathogen concentration tested (0.001% parasitemia). Overall, the automated device (gray bars) extracted less malaria DNA from each sample compared to the manual method (black bars); however, the DNA extracted was detected using PCR at all pathogen concentrations tested.
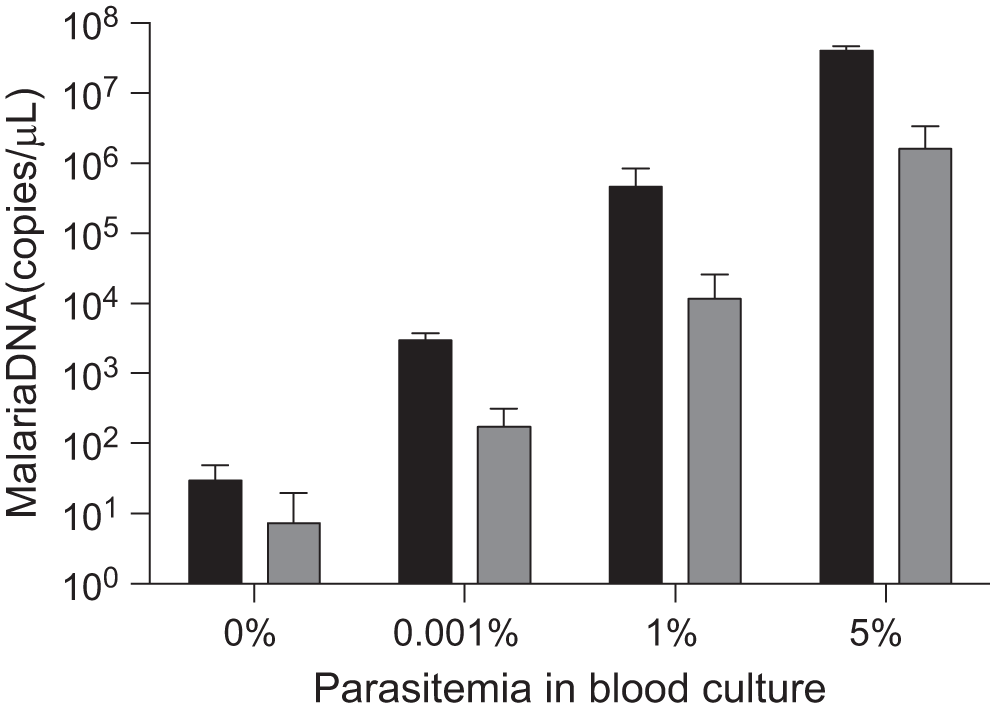
Comparison of manual and two-speed automated extraction methods using Program for Appropriate Technology in Health (PATH)-prepared malaria samples in blood culture. Manual extraction (black bars) performed better than automated extraction (gray bars). The amount of DNA extracted using the automated device was within detection limits of PCR(n = 9, mean ± SD).
The automated biomarker extraction device also performed well using the other PATH-prepared samples, recovering detectable quantities of target biomarker for high, medium, and low levels of infection from each sample matrix ( Fig. 4 ). Results are presented as the concentration of biomarker in the final elution chamber. As expected, the relative amounts of recovered biomarker were proportional to the amount of pathogen present in the samples ( Table 1 ). The concentration of recovered RNA from influenza A–infected swabs was 1.1 ± 0.6 × 105, 2.3 ± 0.4 × 103, and 7.3 ± 2 copies/µL for high, medium, and low levels of infection, respectively ( Fig. 4A ). Approximately 1.8 ± 0.6 × 104, 2.5 ± 0.7 × 103, and 405 ± 48 copies/µL of DNA were detected in high, medium, and low levels of E. coli–infected urine ( Fig. 4B ); 463 ± 472, 69.2 ± 84, and 0.22 ± 0.3 copies/µL in high, medium, and low levels of TB-infected sputum ( Fig. 4C ); and 1.6 ± 2 × 106, 1.2 ± 1.5 × 104, and 167 ± 129 copies/µL in high, medium, and low levels of malaria parasite–infected blood ( Fig. 4D ). Finally, 7.2 ± 2.7 × 103, 512 ± 74, and 2.7 ± 1.9 pM HRPII protein biomarker was detected in high, medium, and low levels of malaria parasite–infected blood ( Fig. 4E ). The PATH samples were prepared as surrogate patient samples using cultured pathogens at known levels of infection. However, the initial pre-extracted concentration of each biomarker target (RNA, DNA, and protein) is not known, because the gene copy number, mRNA levels, and HRPII protein levels are not well established for these pathogens or do not correlate strongly or scale linearly with pathogen infection levels. Therefore, the percentage of the biomarkers recovered after extraction could not be calculated. The results, however, correspond well with the final biomarker concentrations extracted with manual devices in past studies, in which the percentage of biomarker recovered from similarly prepared samples was ~60–80% and approximately equal to commercial gold standard extraction kits.7,9 Taken together, these results indicate that this device extracts clinically important concentrations of DNA, RNA, and protein biomarkers from complex biological samples.
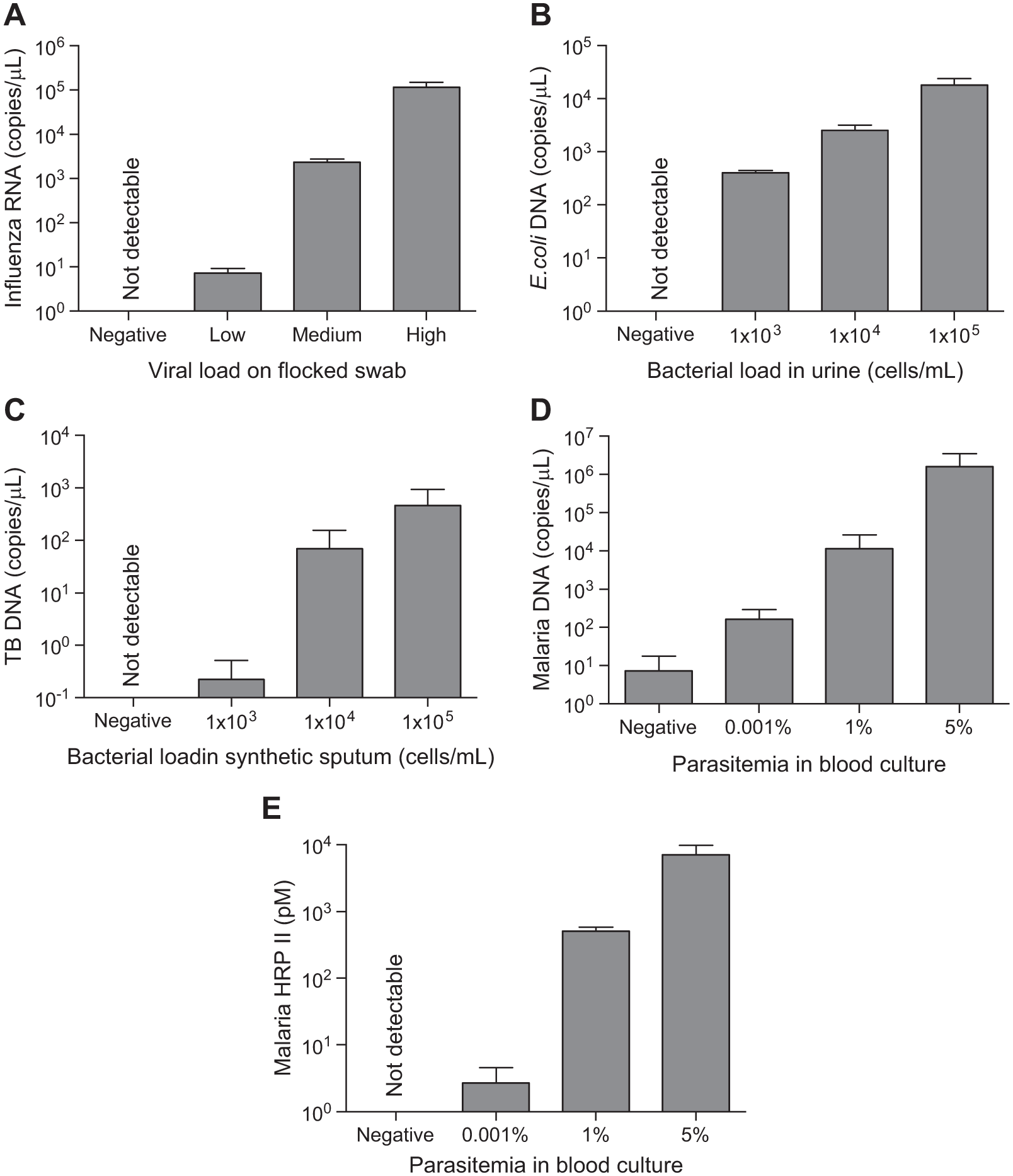
Target biomarkers are detected at biologically relevant high, medium, and low levels of infection for all processed sample panels. All data in A–D are presented on a log scale due to the wide range of biomarker concentrations present at low, medium, and high infection levels. (
There are four advantages to this device that, to our knowledge, have not been incorporated into a single previously reported technology. The first is that this automated biomarker extraction device has the flexibility to extract multiple classes of biomarkers using the same device. The different assay tubes contain different processing solutions and are different lengths, but the physical instrument used to process the tubes is the same in all cases. The user simply has to load an assay tube onto the instrument and choose the correct processing program for the assay. In these studies, the extraction of RNA, DNA, and protein biomarkers was demonstrated using a single device, and it is expected that the automated biomarker extraction device could be used to recover virtually any biomarker classes that could be captured on a surface-functionalized magnetic bead, such as whole cells, lipids, or small molecules. For this reason, this platform represents a significant advantage over other available automated extraction methods that are designed for a single class of biomarker. A second advantage is that the position-agnostic processing program permits random access of assays. This is particularly appealing in settings that receive low sample numbers, because it removes the requirement to batch samples. Users can add or remove individual assay cartridges from the device asynchronously, without stopping or otherwise affecting the processing of other assays. A third advantage of this device is that it improves the sensitivity and general performance of detection methods by concentrating biomarkers and removing background molecules from complex samples. For example, proteins are commonly detected within a complex matrix using ELISA, but we have previously shown that extracting and concentrating proteins using our manual extraction device enhance ELISA and rapid diagnostic test (RDT) detection of low protein concentrations. 9 This indicates that this device can be used to enhance any number of other assays in which low biomarker concentration in the original sample is a concern. A fourth advantage to this automated format is the short extraction time. Magnetic bead processing for each surrogate patient sample was performed in less than 10 min, and the total extraction time, including sample collecting, lysis, and introduction, ranged from 15 to 40 min. Scaling the device to incorporate more than the five extraction cassettes in the current configuration has the potential to increase extraction throughput to the scale required in a clinical laboratory. This is an important consideration when using relatively time-consuming detection schemes such as PCR or ELISA.
We have demonstrated that this automated biomarker extraction device efficiently processes RNA, DNA, or protein biomarkers from surrogate patient samples; however, to make an impact in a clinical laboratory setting, the development of a simplified user interface is needed for the steps before and after sample processing. Currently, loading the assay tube and sample, as well as preparing reagents for PCR or ELISA, requires expensive laboratory equipment, pipetting, and other handling steps that may be expected in a research laboratory but may be too cumbersome to be implemented in a small clinical setting. Future work is focused on streamlining the sample loading procedure and integrating an amplification and detection system into the device. Simplified sample preparation might be achieved by integrating a sample-loading system into each assay cassette to allow sample introduction via a transfer pipette, swab, or other sample collection device. Because the assays developed in these studies each use a slightly different two-step extraction program, the instrument is limited to processing a single biomarker class at a time (i.e., in its current form, the instrument requires the user to change the program for each assay type). Developing a unified two-speed processing protocol that is compatible with each assay type would improve the flexibility of the instrument. Future work with this device will also include integrating optical and thermocycling systems into the instrument and the addition of the amplification and detection reagents into the assay tubes to be able to perform RT-PCR, PCR, and ELISA assays within one complete system. The ultimate aim of these improvements is to develop a clinical device that is fully automated from sample collection to patient diagnosis. This will remove the need for extensive, operator-dependent, sample-handling steps before and after the biomarker has been extracted from the patient sample, allowing untrained technicians to perform a wide variety of diagnostic tests with one instrument.
Conclusions
We have successfully developed an automated biomarker extraction device that isolates multiple biomarker classes (i.e., RNA, DNA, and protein) from clinically relevant surrogate patient samples. The automated format reduces operator-dependent processing steps and increases extraction throughput without requiring complex robotics or fluid pumping. Biomarker recovery using the automated device was comparable to that of the more complex and operator-dependent manual extraction. The continuous-roller format used in this device allows multiple cassettes to be processed simultaneously, and cassettes may be added to or removed from the device without interrupting the processing of other cassettes. This automated device has the potential to simplify and shorten sample preparation for multiple biomarker classes in diagnostic settings.
Footnotes
Acknowledgements
We thank M. F. Richards for critical reading of this manuscript. We are grateful to PATH for providing the surrogate patient sample panels.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Bill & Melinda Gates Foundation through the Grand Challenges in Global Health initiative to FRH and DWW. Support from the National Science Foundation Graduate Research Fellowship Program is gratefully acknowledged by NMA [DGE 0909667] and KMD [DGE 2012095464].