
Abstract
High quantities of quality RNA are necessary for many veterinary laboratory tests. Several commercial kits are available for RNA isolation from human whole blood; their resultant RNA yield and purity have not been reported for canine whole blood, to our knowledge. We assessed the performance of 4 RNA extraction kits (RiboPure, TRIzol, RNeasy Protect animal blood, and QIAamp RNA blood mini). Whole blood from a healthy dog was stored in the manufacturer-recommended RNA stabilizing buffer as directed. RNA isolation, including DNase treatment, was performed using each kit’s manufacturer’s protocol. Resultant RNA yield and purity were evaluated using spectrophotometric absorbance, capillary electrophoresis and electropherogram analysis, and a reverse-transcription real-time PCR (RT-rtPCR) assay. The RNeasy Protect animal blood kit extracted the highest, and RiboPure the lowest, concentration of nucleic acid. RNA integrity numbers classified extracted RNA as good quality or better for all kits except RNeasy Protect. All kits had evidence of genomic DNA contamination as assessed by RT-rtPCR. Overall, QIAamp RNA blood mini kit and TRIzol optimized both RNA yield and purity from canine whole blood. These kits extracted high quantities of good quality RNA as evidenced by high RNA integrity numbers and minimal contamination with proteins and solvents.
Increased use of RNA-based molecular techniques in research and clinical laboratory tests necessitates the extraction of high-quality RNA from whole blood samples in adequate quantities. Increased RNA quantity increases the number of possible downstream applications, expanding the breadth of meaningful gene expression data obtained. Increased extraction quality reduces confounding variables such as genomic DNA (gDNA), DNA-binding proteins, phenol, and other materials that are incompletely separated from RNA. These contaminants can lead to unreliable or misleading results in downstream gene expression analysis. Often, researchers must choose to maximize either RNA quantity or quality, at the expense of the other. Knowing the ability of variable extraction methods to optimize RNA quantity and/or quality is critical when planning for a desired downstream application.
Although there are many commercial kits used to extract RNA from whole blood, there is limited information comparing each technique’s performance regarding RNA purity and yield. Additionally, most RNA extraction kits are designed specifically for use with human whole blood samples. To date, there is one RNA extraction kit that is specifically designed for use with animal whole blood. Canine blood contains less protein and bicarbonate, more chloride, and a higher ratio of sodium-to-potassium compared to human blood. 4 It is unknown whether these differences significantly affect RNA extraction from canine blood when using a human-based whole blood extraction kit.
Given these unknowns, we assessed the performance of 4 commercial RNA extraction kits using canine blood to determine those with optimal RNA yield and RNA purity. The University of Wisconsin–Madison Institutional Animal Care and Use Committee approved our study.
Whole blood was collected from 1 healthy dog for use with 4 kits: RiboPure RNA purification kit (RP; Thermo Fisher), TRIzol reagent (TZ; Thermo Fisher), RNeasy Protect animal blood system (PAB; Qiagen), and QIAamp RNA blood mini kit (RBM; Qiagen). Blood was placed in EDTA anticoagulant tubes, mixed, and aliquoted for all kits except PAB, which used whole, non-anticoagulated blood. RP, TZ, and RBM all utilized 500 μL of anticoagulated whole blood aliquots; PAB utilized 100 μL of blood. Aliquoted samples were then placed in 1 of 3 RNA-stabilizing buffers: RP and RBM in RNAlater (Thermo Fisher), PAB in RNAprotect (Qiagen), and TZ in TRIzol reagent (Thermo Fisher) according to the RNA extraction kits’ recommendations. RBM blood samples had RNA extracted immediately before freezing because the manufacturer stipulates that frozen blood cannot be used. PAB samples were incubated at room temperature for 2 h before being frozen at −30°C; all other samples were frozen immediately (Table 1). Each kit had a minimum of 3 samples frozen for analysis.
RNA extraction kit characteristics including isolation technology, samples and volumes, buffers and volumes, additional processing performed, and cost per run based on prices at time of publication.
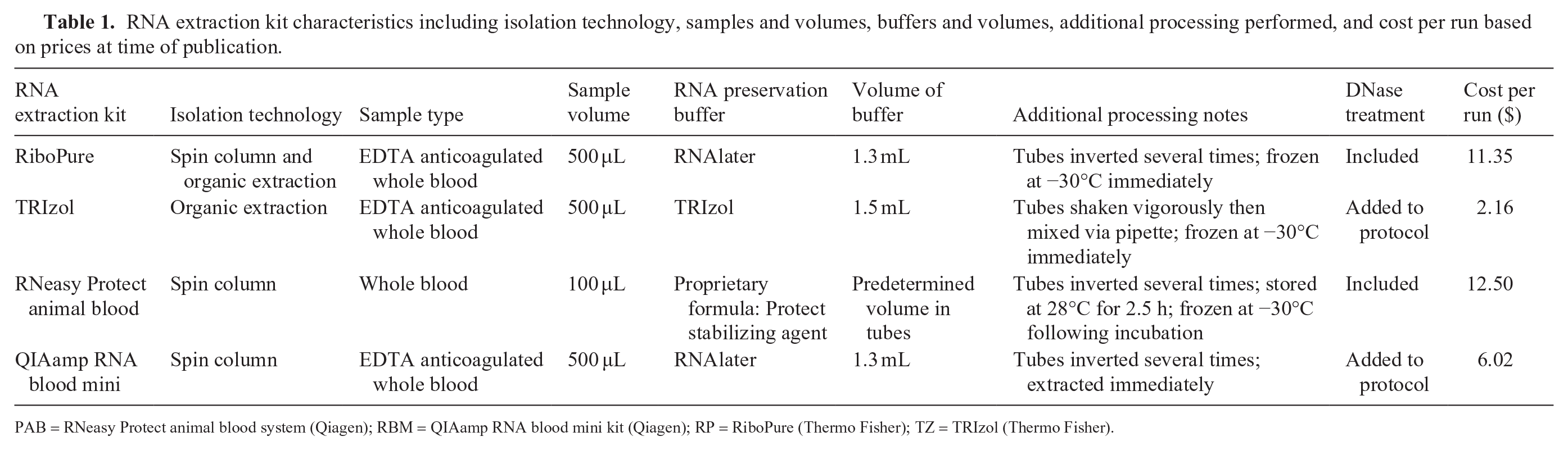
PAB = RNeasy Protect animal blood system (Qiagen); RBM = QIAamp RNA blood mini kit (Qiagen); RP = RiboPure (Thermo Fisher); TZ = TRIzol (Thermo Fisher).
RNA isolation was performed on each sample in triplicate using the manufacturer’s recommended protocol for each kit. RP and PAB kits included DNase treatment steps within their protocols. For TZ and RBM, DNase treatment was performed after RNA isolation using an RNase-free DNase set (Qiagen). All isolated RNA was stored at −30°C.
The RNA yield for each kit was analyzed via spectrophotometric absorbance (NanoDrop 2000 spectrophotometer; Thermo Fisher). RNA purity was assessed with the use of a NanoDrop 2000 and with RNA integrity numbers (RINs) calculated via the 2100 bioanalyzer (Agilent). Using the NanoDrop 2000, contamination with protein or organic compounds was evaluated using the ratio of spectrophotometric absorbance peaks at wavelengths of 260–280 nm (A260/280) and 260–230 nm (A260/230), respectively.
Absence or presence of gDNA contamination was assessed via a reverse-transcription real-time PCR (RT-rtPCR) assay for the reference gene β-actin, using primers constructed to not span introns, thus including gDNA in amplification. The β-actin primer set used was FWD (5’-GACCCTGAAGTACCCCATTGAG-3’) and REV (5’-TTGTAGAAGGTGTGGTGCCAGAT-3’). Complementary DNA (cDNA) was created using TaqMan reverse transcription reagents (Thermo Fisher) per the manufacturer’s protocol, with the exception of a 50-μL reaction volume, extension of the 95°C RT time from 30 to 60 min, and exclusive use of random hexamers. RT-rtPCR was performed on RNA (no-RT control) and cDNA for each sample using 45 cycles of 10 s denaturation at 95°C, 20 s annealing at 62°C, and 20 s extension at 72°C using a LightCycler 96 (Roche). Each PCR reaction for each extraction was completed in duplicate. Cycle threshold (Ct) values obtained from the paired no-RT control and cDNA samples were compared to evaluate gDNA contamination; amplification of a PCR product in the no-RT control samples was considered evidence of gDNA contamination. A single amplification product was confirmed via melt curve analysis.
Statistical analyses were performed using commercial software (Prism 8; GraphPad Software). Data were tested for normality and analyzed using the nonparametric Wilcoxon matched-pairs signed-rank test and Friedman test of repeated measures. Values are reported as means and standard deviations. Statistical significance was obtained with p ≤ 0.05.
RNA quantity was inferred from the total nucleic acid concentration measured via spectrophotometric absorbance (Table 2). PAB extracted the highest concentration of nucleic acid; RP extracted the lowest concentration of nucleic acid. PAB extracted significantly more nucleic acid (p = 0.01) than RP. There was no significant difference in the concentration of nucleic acid extracted by any of the remaining kits assessed.
RNA yield in ng/μL and quality as assessed by capillary electrophoresis and electropherogram analysis.
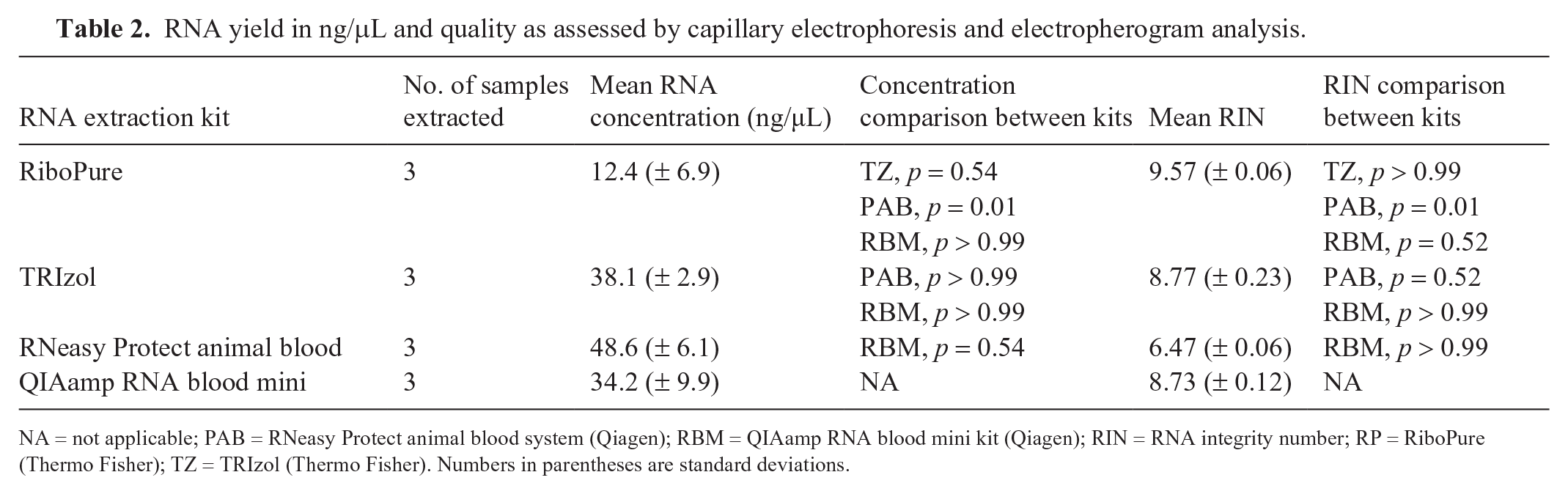
NA = not applicable; PAB = RNeasy Protect animal blood system (Qiagen); RBM = QIAamp RNA blood mini kit (Qiagen); RIN = RNA integrity number; RP = RiboPure (Thermo Fisher); TZ = TRIzol (Thermo Fisher). Numbers in parentheses are standard deviations.
RIN values classified extracted RNA as good quality or better for all kits except PAB (Table 2). RP had a significantly higher RIN than PAB (p = 0.01). There was no significant difference in RIN among RP, TZ, and RBM. Evaluation of electropherograms for all kits showed shorter 28S peaks, indicative of RNA degradation.
In total, 23 of 24 no-RT control PCR runs (n = 6 for each kit, or each of 3 extraction reactions tested in duplicate) had DNA contamination (Table 3), as evidenced by amplification and melt curve indicative of product amplified. RP was the only kit with one sample that had no amplification, indicating minimal gDNA contamination. Additionally, Agilent bioanalyzer electropherograms for PAB were consistent with substantial DNA contamination as evidenced by a lack of return to baseline between 18S and 28S peaks. TZ and RBM had minimal evidence of contamination on electropherogram.
Extract genomic DNA contamination assessment via RT-rtPCR and via spectrophotometric ratios A260:280 and A260:230 to assess presence of DNA, proteins, and other organic compound contaminants.
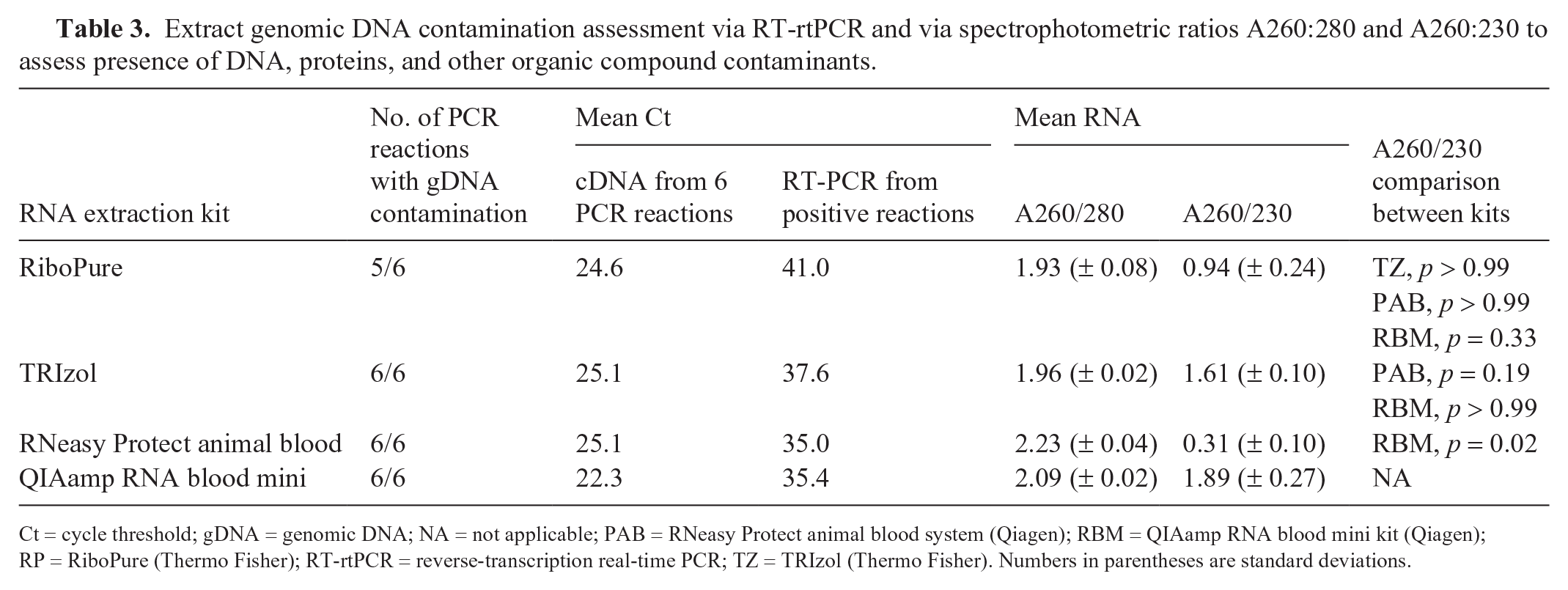
Ct = cycle threshold; gDNA = genomic DNA; NA = not applicable; PAB = RNeasy Protect animal blood system (Qiagen); RBM = QIAamp RNA blood mini kit (Qiagen); RP = RiboPure (Thermo Fisher); RT-rtPCR = reverse-transcription real-time PCR; TZ = TRIzol (Thermo Fisher). Numbers in parentheses are standard deviations.
Contamination with other organic compounds, salts, and proteins was assessed via absorbance wavelength ratios A260/280 and A260/230 via microvolume spectrophotometry analysis (Table 3). There was no significant detection of protein contamination among kits via A260/280 ratios, which were not significantly different between kits. Assessment for other contamination via A260/230 showed significantly more contamination in PAB than RBM (p = 0.02). Consistent with this finding, significant guanidine contamination was found in PAB samples during Agilent bioanalyzer quality control. There was no significant difference in A260/230 among the remaining kits (RBM, TZ, RP).
We identified TZ and RBM as 2 commercial human RNA extraction methods most likely to optimize both RNA yield and purity from canine whole blood samples. Both kits had high quantities of RNA extracted, with good quality evidenced by high RIN and minimal contamination with proteins and solvents. Importantly, for downstream applications such as RT-rtPCR, all kits tested had gDNA contamination despite the included or added DNase steps. Although previous studies have evaluated RNA extraction kits for use with whole blood in several other species, RNA extraction kits have not been evaluated and reported for use with canine whole blood, to our knowledge.3,5,6,9
The differences identified between kits emphasize that researchers must consider their downstream application when choosing extraction kit methodology. For example, although PAB was the only kit marketed as being designed specifically for animal rather than human blood, it exhibited the lowest RIN and the highest evidence of gDNA and other contamination via electropherogram and A260/230. This contamination is likely secondary to the extraction method, given that PAB utilizes spin column technology, which is known to contribute to guanidine contamination and could require additional wash steps on the column. Similar to other studies, the differences in kits studied also emphasize the importance of using a single method throughout the course of a trial or study to minimize RNA quantity or quality confounding variables. 5
Our study used whole blood in storage buffer and not separated peripheral blood mononuclear cells (PBMCs), because we aimed to replicate samples most easily collected in clinical studies. It is known that whole blood contains several PCR inhibitors such as immunoglobulins, lactoferrin, and heme, and abundant globin messenger RNA, all of which could interfere with downstream applications.1,2,9 However, similar to large-scale human clinical trials, veterinary studies may lack the personnel or laboratory space necessary for individual sample PBMC separation or immediate RNA extraction, making whole blood storage more convenient.7,8
As well as the tested RNA yield and purity, there are several additional factors that may influence the choice of an RNA extraction kit. Protocols that utilize organic extraction technology, such as RP and TZ, require more technical skills and expertise than the kits that use spin column technology, such as RBM and PAB. Additionally, the use of phenol and chloroform-based reagents in TZ and RP in laboratories that do not have a chemical fume hood poses a potential safety hazard. Another factor that may influence the selection of extraction kit is cost; in our study, PAB had the highest cost per run but the lowest quality RNA extracted (Table 1).
There are several limitations to our study. First, varying degradation of RNA from RNases throughout the process of whole blood collection and RNA isolation can negatively affect downstream applications and extraction results. 9 In order to minimize this source of experimental error, we took measures to limit exposure to RNases by storing all whole blood in preservation buffers designed to inactivate RNases and retain RNA quality, using certified RNase/DNase-free pipettes and microcentrifuge tubes, and treating surfaces proximal to RNA samples with RNase AWAY reagent (Thermo Fisher). Despite these precautions, all kits assessed had RNA degradation as demonstrated by automated electrophoresis results. Another limitation is the number of kits evaluated and the number of replicates per kit. The kits evaluated were from large manufacturers using recommended protocols; alternative protocols aimed at optimizing kit performance were not evaluated. Testing additional kits, protocols, and more replicates of samples would broaden our understanding of the performance of all commercial RNA extraction kits with canine whole blood. Finally, downstream applications, aside from RT-rtPCR, were not tested with these samples.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.