
Abstract
Nucleic acid biomarkers embody inherent importance for differentiating disease-causing organisms or environmental pathogens. Identifying unknown nucleic acids in low abundance remains extremely challenging. Previously, we reported a method to identify complementary DNA (cDNA) molecules based on sequence-specific topographical labels measured by atomic force microscopy (AFM). However, the accuracy is limited because only one type of nicking endonuclease was used as the labeling agent. Here we investigate how accuracy is improved using multiple types of nicking endonucleases in combinations. The numerical experiments created cDNA molecules incorporating measurement error or labeling defects, which were later compared with the 29,563 human messenger RNA (mRNA) transcript database with ideal labels. After comparison, the unknown cDNA molecule was identified as the transcript with the highest matching score. Thus, the accuracy was determined by the rate of true positives. We found that the accuracy is positively proportional to the label number. Compared with cases using single nicking endonuclease, which has an average accuracy of 51.2% ± 34.4%, the average accuracy was improved to 97.1% ± 5.6% using an optimized combination of NtBsmAI + NtBstNBI + NtAlwI. This improved accuracy is applicable to more than 85% of human mRNA transcripts. Together, our study suggests an optimization strategy for identifying nucleic acids in low abundance using the AFM-based method, with implications for diseases diagnosis, pathogen identification, and forensics at the single molecule level.
Introduction
Nucleic acid sequence represents a unique signature for living organism, including mammals, microorganisms, and virus. It is of particular interest because it often serves as the biomarker associated with diseases or pathogens. For example, emerging evidence has shown that seven signature messenger RNAs (mRNAs) in saliva are elevated due to oral cancer. 1 MicroRNAs, small (~22 nt) regulatory RNAs present in the bloodstream due to the dysfunction of cancer, recently found their promise as tissue-based markers for cancer classification and prognostication. 2 In addition, for analysis of pathogenicity, chromosomal markers with species-specific sequences can be used to differentiate pathogenic strains,3–5 which is important for food safety and homeland security when encountering bioterrorism threats. Thus, effective detection and identification affect many aspects of our lives, including food safety, water contamination, and diseases. 6
There have been many approaches to analyze nucleic acids. Polymerase chain reaction (PCR), coupled with molecular fluorophore assays such as real-time PCR or DNA microarrays, has been widely used for the detection of nucleic acid sequences of interest.5,7–9 In addition, many types of nanomaterials, such as gold nanoparticles, 10 quantum dots,3,11 and electrochemistry,12–14 have been employed for the detection of nucleic acids due to their large surface-to-volume ratio and the unique physical and chemical properties. However, these methods require probes or primers specifically designed to determine the presence of a specific target DNA/RNA. Without a hypothetical target, it is not applicable to differentiate the identity of the unknown nucleic acid species. To identify a DNA/RNA molecule, sequencing methods may be employed, which include Maxam-Gilbert sequencing 15 and chain termination methods 16 for small-scale sequencing and next-generation sequencing, 17 such as the Roche 454 (Roche, Indianapolis, IN), 18 the Illumina/Solexa Genome Analyzer (Illumina, San Diego, CA), 19 and the SOLiD System (Applied Biosystems, Foster City, CA), 20 for large-scale sequencing. However, most methods require cloning to amplify the copy number. The identification of DNA/RNA in low abundance (single molecule level) remains very challenging.
Previously, we reported a method that can identify short complementary DNA (cDNA) molecules based on sequence-specific topographical labeling. 21 The length and label positions of unknown cDNAs can be precisely measured by atomic force microscopy (AFM), which yields a unique labeling pattern for sequence mapping ( Fig. 1 ). Therefore, it offers a new approach to identify nucleic acids without the need of predesigned primers or probes based on a hypothetical target. However, by using only one type of nicking endonuclease, which restricts the number of labeling sites and the population of transcripts suitable for this approach, the identification accuracy is limited.

Schematics of complementary DNA (cDNA) molecules with sequence-specific topographical labels measured by atomic force microscopy.
Here, instead of using only one type of labeling agent, we investigate how accuracy is improved when multiple types of nicking endonucleases are applied simultaneously. We found a positive correlation between accuracy and the number of labels, suggesting a strategy for optimizing the combinations of nicking endonucleases. For cases labeled with only one type of nicking enzyme, the average accuracy is 51.2% ± 34.4%, but with a combination of three selected nicking endonucleases (NtBsmAI + NtBstNBI + NtAlwI), the average accuracy was significantly increased to 97.1 ± 5.6% without substantially increasing the experimental cost and complexity. This improved accuracy is applicable to more than 85% of human mRNA transcripts. Together, our results demonstrate an optimized strategy to identify nucleic acid molecules, with particular importance for applications of samples in low abundance.
Materials and Methods
Simulation of cDNA Molecules
One hundred transcripts were sampled from 29,563 human mRNA transcripts from the NCBI Reference Sequence (RefSeq) database based on their length distribution ( Fig. 2 ). Eight types of nicking endonucleases (NtBspQI, NtBbvCI, NbBsmI, NbBsrDI, NbBtsI, NtAlwI, NtBstNBI, and NtBsmAI) were used in different combinations for labeling the sampled transcripts. Based on each sampled transcript and a specific combination of nicking endonucleases, 100 cDNA molecules were generated, incorporating the errors in labeling and AFM measurement. The AFM measurement is limited by its lateral resolution of 5 nm. As such, the measurement error of labeling position was defined in a range of normal distributed deviations from the ideal labeling site, and 5 nm was set as the standard deviation of the normal distribution. Considering incomplete reverse transcription when generating the cDNA molecules, we implemented a truncation of 0% to 20% of length from the 5′ end, which was uniformly distributed among the 100 cDNA molecules. For missing labeling, the probability was assumed as 20% per site. For spurious false labeling, which means labeling to a wrong site randomly, the probability was assumed as 3%.
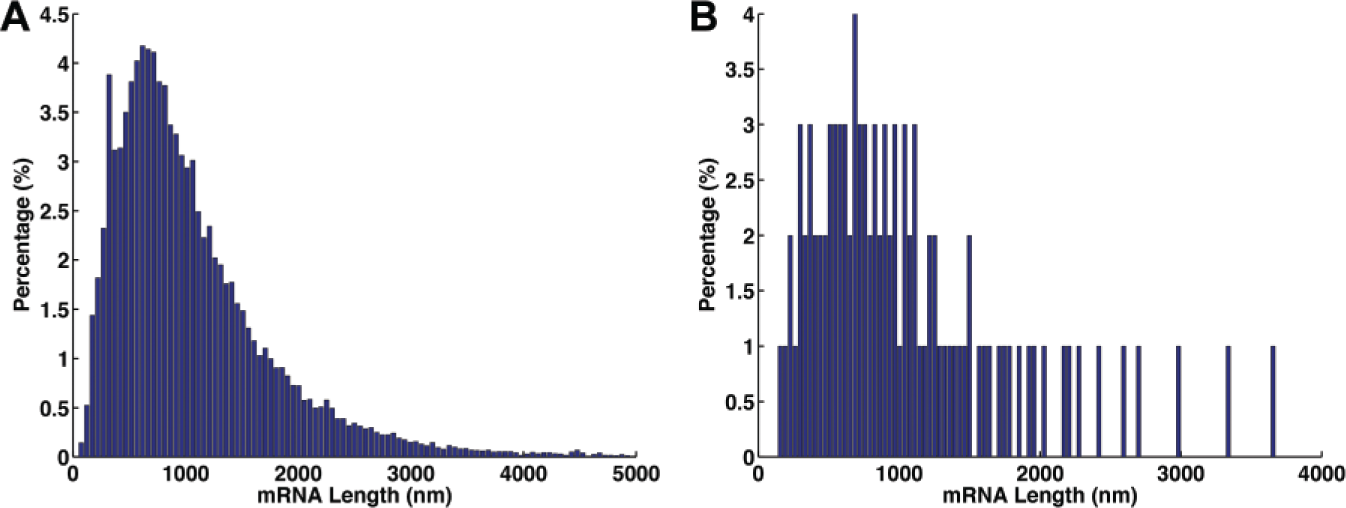
Distribution of human messenger RNA (mRNA) transcripts. (
Matching the 100 cDNA Molecules to the Library with Ideal Labeling Patterns from the Human mRNA Transcripts Database
Each simulated cDNA molecule was compared with the 29,563 human mRNA transcripts from the NCBI RefSeq database. Using a specific combination of nicking endonucleases, the 29,563 mRNA transcripts were labeled with the nicking endonucleases without considering the errors, which renders a library of ideal labeling patterns. Based on the library, each cDNA molecule was aligned with each transcript decorated with the ideal labeling pattern from the 3′ end (
Results
Principle of the Sequence-Specific Topographical Labeling for Identifying cDNA Molecules
This principle is based on our previously reported method, sequence-specific topographical labeling. 21 Before AFM scanning, cDNA molecules of unknown sequence were labeled with nicking endonucleases that can recognize specific sequences of five to seven nucleotides (e.g., Nt.BspQI for 5GCTCTTCN3). Subsequently, the nicked cDNA molecule was enzymatically modified with biotinylated nucleotides, allowing bindings of streptavidin at the labeling sites and making them visible by AFM ( Fig. 1 ). Note that multiple sites in a single cDNA molecule can be simultaneously labeled. As such, based on the labeling positions, the spacing between the labeling, and the total length of the cDNA molecule, this labeling pattern yielded unique characteristics representing the unknown cDNA molecule. Consequently, comparing the measured labeling pattern with a library of ideal labeling patterns of known mRNA transcripts, the transcript with the highest matching score was used as the identity of the unknown cDNA molecule, providing a measurement at the single molecule level.
Generation of cDNA Molecules That Underwent Sequence-Specific Labeling
To evaluate the accuracy of this principle, we first conducted numerical experiments to simulate the cDNA molecules that underwent the sequence-specific labeling. Specifically, we simulated the labeling of the molecules considering the experimental errors, which may occur in biochemical reactions or AFM imaging. Eight types of nicking endonucleases (NtBspQI, NtBbvCI, NbBsmI, NbBsrDI, NbBtsI, NtAlwI, NtBstNBI, and NtBsmAI) were applied in different combinations for these numerical experiments. The NCBI RefSeq database of 29,563 full-length human transcripts was used as the model library. On the basis of the mRNA lengths of the entire library (
Fig. 2A
), we sampled 100 transcripts representing the general mRNA population (
Fig. 2B
). For each sampled transcript pairing with one specific combination, 100 cDNA molecules were generated incorporating the following hypothetical errors: (1) inaccurate measurement of labeling position by AFM, (2) truncation at the 5′ end due to incomplete reverse transcription, (3) missed labeling, and (4) spurious false labeling. An example of 100 cDNAs generated from the OR8B2 mRNA transcript labeled with a combination of NtBstNBI, NtBsmAI, and NtAlwI is shown in
Accuracy of the Sequence-Specific Topographical Labeling for Identifying cDNA Molecules
On the basis of the 100 simulated cDNA molecules representing one sampled transcript labeled with a specific combination of nicking endonucleases, we next evaluated the accuracy of such sequence-specific topographical labeling for identifying the cDNA molecules. The labeling patterns of the 100 simulated cDNA molecules were compared with the ideal labeling patterns of the 29,563 human mRNA transcripts from the NCBI RefSeq database. We defined a true positive if the simulated cDNA molecules and its corresponding transcript with ideal labeling had the highest matching score. In contrast, if the simulated cDNA molecule was paired with an irrelevant transcript because of the higher matching score, the alignment was defined as false positive. Thus, the accuracy was calculated as the percentage of true positives among the 100 cDNA molecules. By placing the accuracy from all 100 sampled transcripts applied with all possible combinations of eight nicking endonucleases together (~28 combinations × 100 cDNA molecules for each sampled transcript; total 100 sampled transcripts), the results show that there is a clear increase of accuracy when the label number of each molecule is increased ( Fig. 3 ). For a label number no less than 10, the accuracy reaches 80% to 100% from different combinations of nicking endonucleases. Moreover, we also found the accuracy is increased when the mRNA transcript is longer (color map in Fig. 3 ). This is because for longer transcripts, there are fewer mRNAs in the library that have comparable length ( Fig. 2A ), therefore minimizing the potential mistake in pairing.
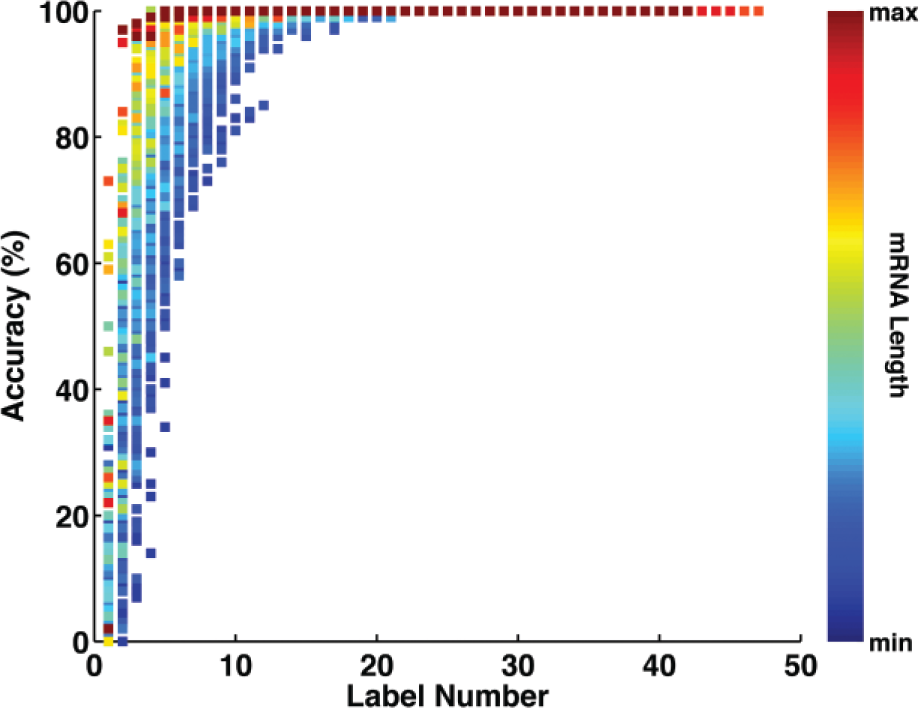
Accuracy of identifying the 100 sampled messenger RNA (mRNA) transcripts using different combinations of nicking endonucleases. The identification accuracy manifests a positive correlation with the label number and the mRNA length.
Label Number of the Eight Types of Nicking Endonucleases
As the label number has strong influence on the accuracy, we next studied the label numbers for each type of nicking endonuclease applied to the entire 29,563 full-length human mRNA transcript library. According to the numerical experiments on the 100 sampled transcripts, accuracy is favored by having an increased label number ( Fig. 3 ), which implies that more types of nicking endonucleases should be applied simultaneously. However, using many types of nicking endonucleases is impractical because it increases cost and experimental complexity. Thus, an optimized strategy should be sought to balance the use of nicking endonucleases and at the same time maintain the overall accuracy at a sufficient level. Based on the entire library, the label numbers of each type of nicking endonuclease show that NtAlwI, NtBstNBI, and NtBsmAI provide the most label numbers compared with the other five types ( Fig. 4A ), which also can be seen by the average label numbers of them ( Fig. 4B ). Interestingly, compared with other nicking endonucleases, the recognition sites for NtAlwI, NtBstNBI, and NtBsmAI are the shortest, only five nucleotides ( Fig. 4B ), suggesting that the shorter recognition sequence has a higher chance to create labels.
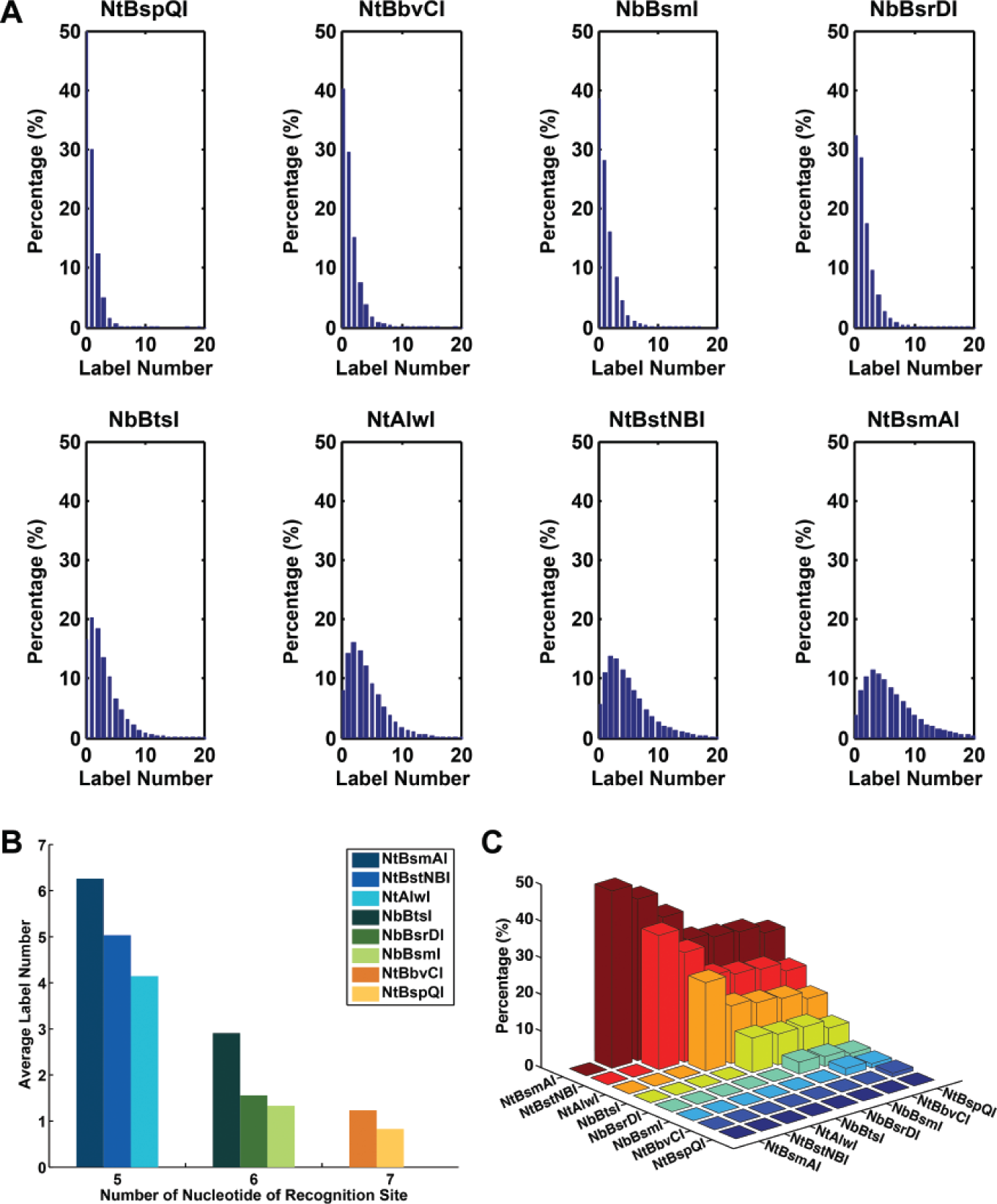
The analysis of the label numbers for each nicking endonuclease. (
Optimized Combination of Nicking Endonucleases for the Entire Transcript Library
We next sought an optimized combination of nicking endonucleases to improve the overall accuracy. Among the total eight types of nicking endonucleases, we first studied the combinations that use only two. A label number no less than 10 was chosen as the threshold since it offers accuracy between 80% and 100% for all 100 sampled transcripts from different combinations of nicking endonucleases ( Fig. 3 ). For the total 28 (8C2 “8 choose 2”) combinations of nicking endonucleases against 29,563 full-length human transcripts, we found that by using a combination of NtBsmAI + NtBstNBI, there are 48.4% of total transcripts having a label number no less than 10, which is the best among the 28 possible combinations ( Fig. 4C ). This result is consistent with the aforementioned findings that NtBsmAI and NtBstNBI have the highest average label numbers because of their short labeling sites ( Fig. 4B ), therefore increasing the label number as well as the population of transcripts having a label number no less than 10. More important, among the 100 sampled transcripts, the average accuracy of 99.5% ± 1.2% (mean ± SD; from 94%-100%) was achieved for cases labeled by NtBsmAI + NtBstNBI and with a label number no less than 10. Of note, for cases that used only one of the eight types of nicking endonuclease, the accuracy is only 51.2% ± 34.4% (mean ± SD; from 0%-100%). It suggests a significant improvement using the combination of nicking endonucleases.
For convenience, cumulative distribution of label number was used to study the population of human mRNA transcripts and their label number. The cumulative distribution was plotted by the percentage of human mRNA transcripts (y-axis) that had no less than a specific value of label number (x-axis). Thus, for the combination of NtBsmAI + NtBstNBI, we can easily determine 48.4% when the label number was no less than 10 ( Fig. 5A ), which corresponds to 99.5% ± 1.2% accuracy.
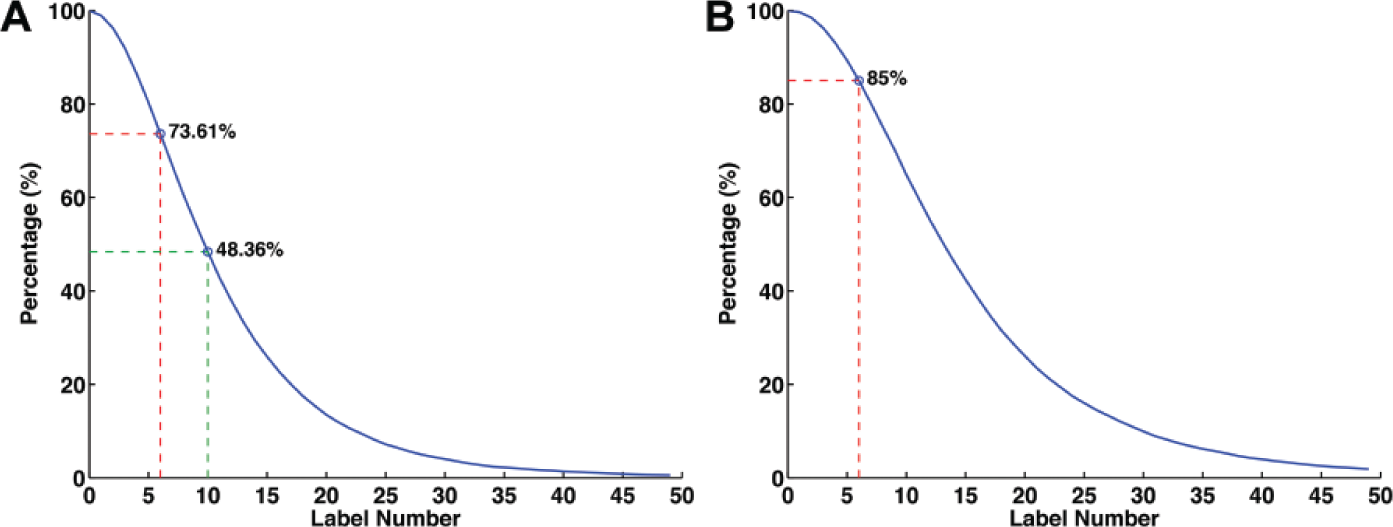
Cumulative distribution of the percentage of human messenger RNA transcripts that has no less than a specific value of label number (
We next explored whether it is possible to enlarge the population of human mRNA transcripts applicable for this improvement. The threshold of a label number, 10, was chosen during the analysis above. However, the population of mRNAs that has a label number no less than 10 was only 48.4%, restricting the applicable range of this optimized combination. To enlarge the applicable range, we first changed the threshold of label number to 6. The results showed that, while the accuracy was slightly decreased to 96.4% ± 6.6% (mean ± SD; from 73%-100%), the applicable range was significantly enlarged from 48.4% to 73.6% ( Fig. 5A ) because more mRNAs have a label number no less than 6.
To further improve the accuracy as well as the applicable range, we resorted to a combination of three nicking endonucleases. NtBsmAI + NtBstNBI + NtAlwI was chosen because they used the shortest recognition sites and had the most average label numbers ( Fig. 4B ). The cumulative distribution shows that 85% of human mRNA transcripts had label number no less than 6 using this combination ( Fig. 5B ). More important, the average accuracy was improved to 97.1% ± 5.6% (mean ± SD; from 76%-100%) for cases labeled by NtBsmAI + NtBstNBI + NtAlwI and with a label number no less than 6. Consequently, based on our numerical analysis, by using the combination of nicking endonucleases, NtBsmAI + NtBstNBI + NtAlwI, in this sequence-specific topographical labeling, an average accuracy of >97% was achieved for 85% of human mRNA transcripts.
Discussion
Nucleic acids represents important biomarker for many diseases and pathogens, especially for applications that require accurate estimation of the levels of target molecules in low abundance (~100), such as patient status or environmental contamination. In this article, we demonstrate that, based on the sequence-specific topographical labeling, the average accuracy was improved from 51.2% ± 34.4% using single nicking endonuclease to >97% by using combinatory nicking endonucleases. According to our numerical experiments, this improved accuracy can be applicable for more than 85% of human mRNA transcripts. Moreover, instead of using eight types of nicking endonucleases, this improvement can be obtained using only three types of nicking endonucleases with shortest recognition sites; therefore, accuracy was improved without sacrificing the experimental cost and complexity.
In this study, we found that accuracy is positively correlated with the label number ( Fig. 3 ). As the labeling pattern is the key characteristic to identify the unknown cDNA molecule, which includes the location of and spacing between the labeling sites on cDNA molecules, more labeling sites will definitely provide more informative and unambiguous labeling patterns for matching with the mRNA transcript library. As such, although combinations of nicking endonucleases are used, it may not be necessary to distinguish which type of nicking endonuclease is at each of the labeling sites as long as the label number is sufficiently high.
Interestingly, we have found that the accuracy also has a positive correlation with the mRNA length, even if the label number is the same (color map in Fig. 3 ). We propose two reasons for this. First, the population of long mRNA transcripts is small, according to the distribution of the mRNA population ( Fig. 2A ), thereby decreasing the probability of mismatching. Second, the measurement error by AFM, which was defined as 5 nm in our simulations, is more dominant in shorter cDNA, which may result in a larger probability of mismatching.
This AFM-based method has demonstrated effectiveness in identifying transcript isoforms previously. 21 However, it may have limitations in differentiating cDNA molecules with single-base mutations. As the typical lengths of nicking endonuclease recognition sequences are five to seven nucleotides, most of the unlabeled parts are only providing information about the spacing between the labeling positions and the total length of the cDNA molecules. As a result, it may be difficult to differentiate the single-base mutation if it occurs at the unlabeled parts. Thus, while this method provides a highly accurate identification of cDNA with unknown sequence, postscreening for the single-base mutations 22 may be needed for the detection of single-base or point mutations.
Together, our results demonstrate an identification of unknown cDNA molecules in low abundance. Importantly, this method does not rely on probes or primers designed to determine the presence of a specific target DNA/RNA, providing a new analytical strategy for disease diagnosis, pathogen identification, and forensics at the single molecule level.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We are pleased to acknowledge the support from the National Natural Science Foundation of China (grant no. 51305375) and the Croucher Foundation (grant no. 9500016).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.