
Abstract
Introduction:
Nucleic acid synthesis is a dual-use technology that can benefit fields such as biology, medicine, and information storage. However, synthetic nucleic acids could also potentially be used negligently and ultimately cause harm, or be used with malicious intent to cause harm. Thus, this technology needs to be appropriately safeguarded. Sequence screening is one component of a biosecurity protocol for preventing such harm and consists of identifying Sequences of Concern (SOCs). There exist many fit-for-purpose tools that have been developed for nucleic acid synthesis sequence screening. However, questions remain regarding their performance with respect to the consistency of screening.
Methods:
To aid in determining if screening tools are harmonized in regard to baseline sequence screening (which represents a minimum acceptable level of performance), the National Institute of Standards and Technology (NIST) constructed a test dataset based on current screening recommendations. NIST then sent blinded datasets to sequence screening tool developers for testing.
Results:
Overall, there was a general agreement between the tools and NIST labels given to the sequences, and all tools had a baseline performance of >95% sensitivity and >97% accuracy. Disagreement on specific sequences largely arose from single tools and could be traced to differences in defining a SOC and/or methodological differences in screening algorithms.
Keywords
Introduction
Nucleic acid (NA) synthesis technology is largely considered a “dual-use” technology. It can be used for the good of humanity, but could have destructive potential if its use is unguarded. 1 NA synthesis has been instrumental in understanding the basic genomic elements of life through the construction of a minimal bacterial genome. 2 It has also been used for disease diagnostics, 3 crop improvement, 4 and in biomanufacturing of biofuels. 5 Furthermore, in the future, NA synthesis may potentially be used for storing information. 6 The capabilities of this technology, which underpins vast domains of biological research, continue to improve, 7 alongside the molecular biology tools for manipulation and assembly of synthetic NA. As NA synthesis becomes more accessible, however, the number of actors who could potentially misuse it to create or enhance pathogenic microorganisms grows. For example, many were unsettled by the synthesis of horsepox virus, 8 concerned that it might lower barriers to creating viruses of pandemic potential. 9
One way to combat the potential misuse of NA synthesis technology is for synthetic NA providers to screen NA synthesis orders for Sequences of Concern (SOCs) that could be used to cause significant harm. In 2010, the U.S. Department of Health and Human Services (HHS) released guidance for synthesis screening titled, “Screening Framework Guidance for Providers of Synthetic Double-Stranded DNA.” While some of the guidance dealt with customer screening, it also outlined details on sequence screening and record keeping. 10 The guidance suggested that all double-stranded DNA (dsDNA) orders (regardless of length) be screened for similarity to sequences from pathogens and toxins on the Biological Select Agents and Toxins (BSAT) list, and for international orders to also be screened for similarity to the Commerce Control List (CCL). 10 The main goal of sequence screening is thus to distinguish SOCs from non-SOC sequences. The initial scope delineated SOCs from non-SOCs based on similarity to sequences from federally regulated agents or toxins. A “best match” approach was recommended whereby synthesis providers would screen all possible 200 bp windows (and all six open reading frames) in an ordered nucleotide sequence to see if the greatest percent identity match was to a regulated agent and thus deemed a SOC. The guidance also supported the use of screening methodologies that were equivalent or superior to the “best match” approach, along with the formation of custom curated databases. 10 Identifying a sequence of concern in an order would then prompt further screening for customer legitimacy.
Around the same time (2009) as the initial U.S. guidance was introduced, the International Gene Synthesis Consortium (IGSC) was formed with the goal of promoting both the beneficial uses of DNA synthesis technology along with appropriate biosecurity measures to mitigate potential risks.11,12 The consortium was focused on five areas (sequence screening, customer screening, record keeping, order refusal/reporting, and regulatory compliance). 13 In regard to sequence screening, IGSC members screen orders against a custom curated Restricted Pathogen Database as outlined in the IGSC’s Harmonized Screening Protocol. 14 This database includes sequences from the aforementioned lists (BSAT and CCL) of regulated sequences. Since its initial founding with five members, the IGSC has grown to include 34 members. 13
In 2023, HHS issued an updated guidance document, “Screening Framework Guidance for Providers and Users of Synthetic Nucleic Acids.” 15 Key updates pertaining to sequence screening as compared with the prior (2010) guidance include: (1) expanding sequence screening to include single-stranded and double-stranded DNA and RNA; (2) implementing a lower limit for screening of 50 nucleotides (nt) by October 2026, across all frames; (3) expanding the definition of SOC to include all sequences known to contribute to pathogenicity or toxicity, “as soon as it is practicable to do so”; (4) considering the ability of shorter SOCs to be assembled from bulk orders or multiple orders over time if overlaps are present; and (5) applying integrated SOC screening into benchtop NA synthesizers. 15
In 2024, the White House Office of Science and Technology Policy (OSTP) released a “Framework for Nucleic Acid Synthesis Screening,” which incorporated and supplemented portions of the 2023 HHS Guidance. 16 Notably, the 2024 OSTP Framework required that synthetic NAs purchased with Federal funds must be obtained from providers or manufacturers that adhered to the Framework via self-attestation (first-party conformity assessment). In 2025, the White House issued Executive Order 14292, “Improving the Safety and Security of Biological Research,” which requires the revision or replacement of the 2024 OSTP Framework with an updated Framework that incorporates enforcement mechanisms to ensure compliance. 17 It is likely that this revised Framework will be even more stringent in regard to conformity assessment than the one released in 2024.
Irrespective of the conformity assessment parties, standards are necessary and should specify all essential characteristics for achieving the objective of the conformity assessment. To establish tools and capabilities for testing sequence screening tools, the National Institute of Standards and Technology (NIST) initiated an effort to develop robust standards for NA synthesis order screening. This included the creation of datasets that can be utilized by sequence synthesis providers to demonstrate baseline sequence screening capabilities, where “baseline” represents a minimum acceptable level of performance. We note that an important and evolving need is methods for validating the expanded SOC definition, which is beyond the scope of the current study.
As NA synthesis capabilities expand, screening requires more sophisticated computational algorithms alongside custom databases. Many providers have thus come to rely on fit-for-purpose tools developed and maintained specifically for NA synthesis sequence screening, which can outperform more generic tools and databases. 18 Some tools currently available include Aclid, 19 The Common Mechanism, 20 FAST-NA Scanner, 21 SeqScreen, 22 SecureDNA, 23 and UltraSEQ.24,25 While each tool has a main goal of detecting SOCs in NA synthesis orders, they employ different search algorithms and databases (Table 1).
Description of sequence screening tools listed in alphabetical order
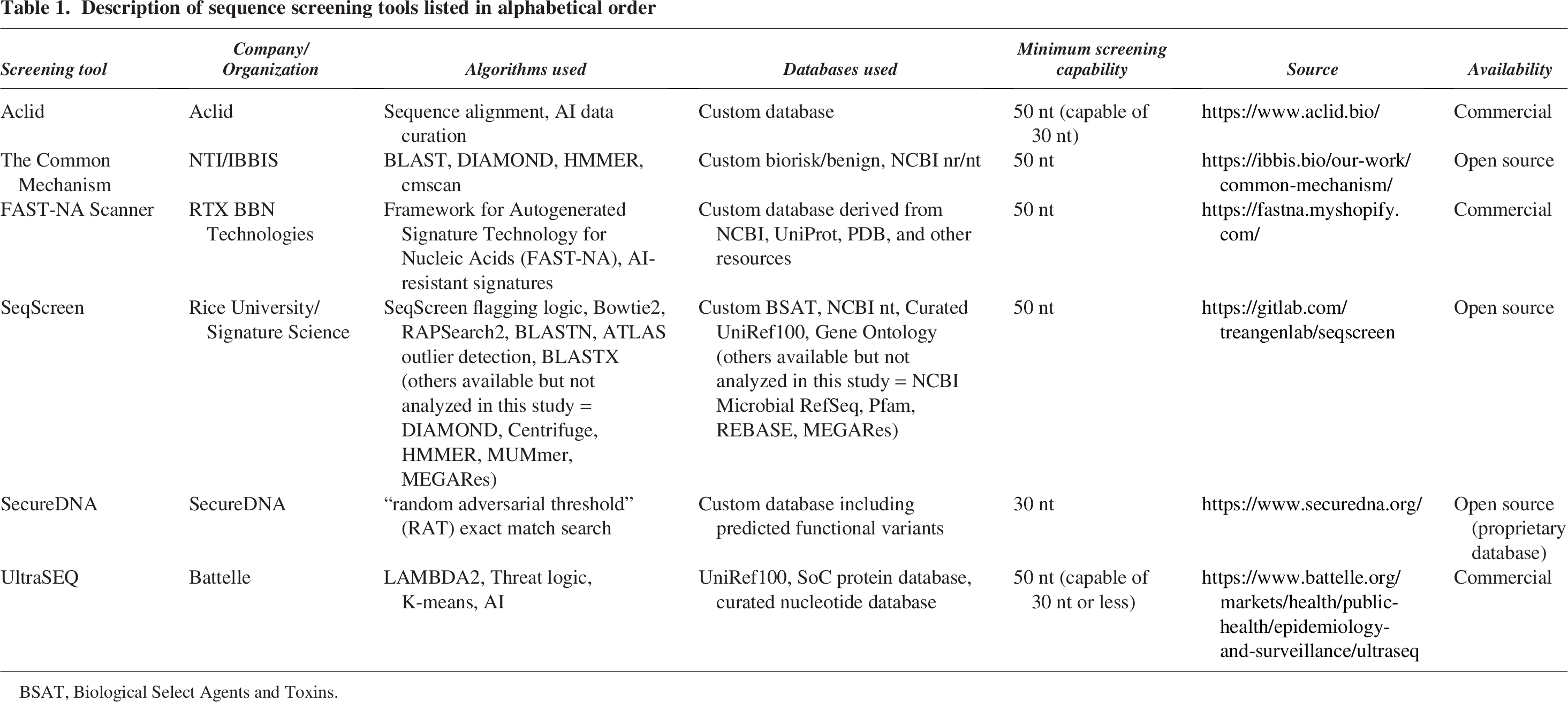
BSAT, Biological Select Agents and Toxins.
In the process of constructing datasets that NA synthesis providers could use to demonstrate baseline sequence screening, NIST sought feedback from developers of the aforementioned tools. NIST sent a blinded test dataset to four tool developers (Aclid, Battelle, IBBIS, and RTX BBN Technologies) to assess its utility and obtain feedback. In addition, NIST ran the same dataset through two additional tools, one that was installed locally (SeqScreen, 22 created by Rice University and Signature Science) and another that utilized a locally installed client to make API calls to a remote database (SecureDNA 26 ). Since the goal was to construct datasets consisting of SOCs and non-SOCs, this assessment with tool developers allowed NIST to gauge whether the industry and government interpretation of the 2023 HHS guidance were in agreement. This article discusses the construction of the test dataset and the baseline performance of the various tools using that dataset. Overall, we found the dataset, as constructed, to be useful in demonstrating baseline sequence screening. In addition, all tools performed well in identifying SOCs (i.e., they have high sensitivity) and most of the disagreed-upon sequences came about due to slightly different definitional interpretations of the HHS guidance.
Methods
Dataset Construction
Annotated virulence factors of bacterial and viral origin were obtained and labeled as SOCs/“TP” sequences in our dataset. For this, bacterial virulence factor genes from organisms on the BSAT list were obtained from the VFDB set A (core) nucleotide sequences. A blastn 40 search of these nucleotide sequences against a locally constructed database of complete RefSeq genomes (n = 62094) was performed. The results were parsed to identify 200 nt windows that were unique to BSAT organisms within the scope of this local database. These sequences were deemed bacterial TPs. Viral virulence factors were obtained by querying the UniProt database for entries that matched the GO term GO:0019049 (virus-mediated perturbation of host defense response) and that belonged to one of the BSAT viral taxa (based on NCBI taxonomy id). These UniProt IDs were then mapped to genomic coding sequences in the NCBI nucleotide database (when available), and FASTA nucleotide sequences were obtained. The resulting sequences were clustered at 100% identity using the program CD-HIT 41 to remove redundant entries that occur due to viral polyproteins. A blastn search of these nucleotide sequences was performed against the same locally constructed database of complete RefSeq genomes as above (n = 62094). SNPs were identified on 200 nt windows with a 50 nt step size in order to obtain sequences unique to BSAT viral genomes. The pool of sequences was randomly downsampled to 250 bacterial TPs and 250 viral TPs. While these 500 TP sequences were all tested, upon later inspection, one bacterial sequence was found to be well below the 200 nt threshold, and we omitted it from further analysis.
TN sequences (non-SOCs) were obtained by randomly selecting 50 fragments, each 200 nt in length, from five bacterial and five viral (phage) genomes that were from non-BSAT organisms that are not known human pathogens. Bacterial sequences came from the genomes of Lactobacillus acidophilus La-14, Bifidobacterium breve DSM 20213, E. coli str. K-12 substr. MG1655, Bradyrhizobium diazoefficiens USDA 110, and Bacillus amyloliquefaciens IT-45. Viral sequences came from the genomes of Escherichia phage T7, Inovirus M13, Microviridae phi-CA82, Leuconostoc phage Ln-7, and Geobacillus phage TP-84.
The TN sequences (non-SOCs) were combined with the TP sequences (SOCs) in a randomized order and given anonymized headers. The combined and anonymized dataset was then tested by each tool developer.
Study Design
The sequences in the dataset were combined, shuffled in order, and blinded using unique numeric identifiers. This blinded dataset was then sent to the tool developers of Aclid, The Common Mechanism, FAST-NA Scanner, and UltraSEQ. Results were then sent back to NIST, where they were unblinded and analyzed. Detailed analysis reports were sent to each tool developer to facilitate feedback and discussion about the utility of the dataset.
The same blinded dataset was input into one locally installed tool at NIST (SeqScreen) and another, which utilized a locally installed client to make API calls to a remote database (SecureDNA). Unblinded results and analysis reports were sent to each respective tool developer, similar to above.
Tool Settings
SecureDNA
The SecureDNA interface software, Synthclient, was run through the command line, and individual sequence queries were sent via the available API using a Python script on July 23, 2024, with the U.S. flagging option selected.
SeqScreen
SeqScreen (version 4.5) was run in a conda environment and executed to run in sensitive mode against the SeqScreen23.4 database using the command: “seqscreen --fasta SoC_test_dataset_0.2_blinded.fa --sensitive --databases SeqScreenDB_23.4 --working SeqScreen_results --threads 30”. The “flag” column was used as a binary indicator of “Flag” versus “No Flag.”
Common mechanism
The multiple columns of the Common Mechanism results file were parsed in order to identify binary “Flag” versus “No Flag” classifications. “No Flag” sequences were defined using the query: ‘biorisk == “P” and regulated_virus == “P” and regulated_bacteria == “P” and regulated_eukaryote == “P” and benign == “-” and mixed_regulated_and_non_reg == “P”’.
UltraSEQ
UltraSEQ settings were used as described in Gemler et al. 25 Any sequence that was classified as Risk Level 1, 2, 3, or 4 was flagged. Risk Levels 5 and 6 were considered “No Flag” for this test.
Aclid and FAST-NA scanner
Aclid and FAST-NA Scanner provided results according to their default settings for current commercial deployments and reported “Flag” and “No Flag” Boolean results.
Performance Metrics
Sensitivity was defined as TPs/(TPs + FNs), where TPs were sequences that were designated to be flagged (SOCs) in the test dataset. Accuracy was defined as (TPs + TNs)/(total number of sequences), where TNs were sequences designated as non-SOCs within the test dataset. The FN rate was calculated as 1 − sensitivity. The FP rate was calculated as FPs/(TNs + FPs). Specificity was calculated as TNs/(FPs + TNs). The NPV at a 5% prevalence of SOCs was calculated as specificity × 0.95/(specificity × 0.95 + [1 – sensitivity] × 0.05).
Examination of Disagreed-Upon Sequences
Sequences were subjected to blastn and blastx searches using the NCBI nt/nr databases in order to identify algorithmic and database discrepancies with NIST labels. We also held follow-up conversations with each tool developer to discuss the result of each tool’s performance and better understand discrepancies.
Power Analysis for Dataset Implementation
Statistical power for sensitivity was calculated using the following function from the R package “pwr” 42 : pwr.p.test(h=ES.h(p1 = 0.95, p2 = 0.984), n = 200, sig.level = 0.05, alternative = “greater”) which resulted in a power value of 0.87. To calculate effect size with the nested function: “ES.h(),” the value for p1 of 0.95 corresponds to the proposed sensitivity cutoff of 95% and the value for p2 of 0.984 corresponds to the mean sensitivity value of the algorithms tested in this article. The value for n is 200 to correspond with the number of graded TP sequences in the datasets for implementation with providers. The power calculation for accuracy was done using the same function with different parameter values: pwr.p.test(h=ES.h(p1 = 0.5, p2 = 0.75), n = 400, sig.level = 0.05, alternative = “greater”), which resulted in a power value of 1.0. The value for p1 of 0.5 corresponds to an accuracy due to random choice or a case where all test sequences are flagged as TPs, and the value of p2 corresponds to the proposed 75% accuracy threshold. The value for n is 400 to correspond with the total number of graded sequences in the datasets for implementation.
Results
Test Dataset Construction
A set of ∼10,000 true positive (TP) sequence fragments (each 200 nt long) was generated by starting with functionally pathogenic bacterial and viral genes from several publicly available databases. Bacterial genes were obtained from the Virulence Factor Database (VFDB), 27 while viral genes were obtained from UniProt 28 with the Gene Ontology (GO) term “virus-mediated perturbation of host defense response” (GO:0019049). These genes were aligned across all complete genomes in RefSeq to identify 200 nt gene fragments that were unique to the regulated organisms on the BSAT list 29 based on their NCBI taxonomy ID. True negative (TN) sequences (non-SOCs) were generated by randomly selecting 50 fragments, each 200 nt in length, from five bacterial and five viral (phage) genomes in RefSeq that were from non-BSAT organisms that are not known human pathogens. While non-BSAT organisms that are known pathogens represent another relevant category of organisms whose genomes contain SOCs, this present study focused on the current (pre-2026) scope for defining SOCs.
A test dataset was then constructed by randomly sampling 249 bacterial TPs and 250 viral TPs, and combining them with the 250 bacterial TNs and 250 viral TNs (Table 2) (one bacterial TP sequence was removed for quality control reasons, see the Methods section for details). The 999 sequence fragments were deidentified and evaluated across six currently available sequence screening algorithms (Aclid, The Common Mechanism, FAST-NA Scanner, SeqScreen, SecureDNA, and UltraSEQ). Sequence screening results were analyzed by NIST and discussed post hoc with the algorithm developers to understand performance.
Dataset overview
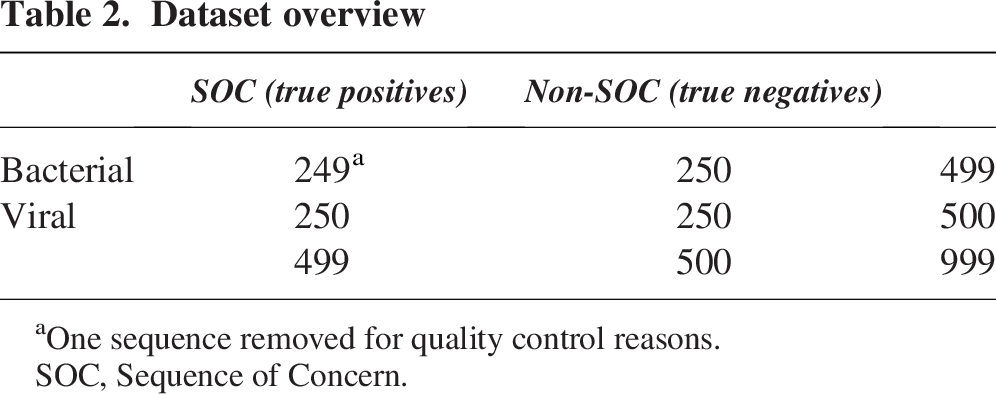
One sequence removed for quality control reasons.
SOC, Sequence of Concern.
Test Dataset Validation
Majority agrees with test dataset
We first ran the entire test dataset through all six tools. The tools classify each sequence as a “SOC” or “non-SOC” which together we refer to as “calls” throughout this report. Then, we compared the calls from the sequence screening tools to the TP (SOC) or TN (non-SOC) designations given to the sequences, which we refer to as the NIST labels throughout this report.
In total, running the test set of 999 sequences through six tools yielded 5994 calls that could be compared with the NIST labels. Our analysis revealed that 46 of these calls, corresponding to 43 unique sequences, resulted in a false negative (FN) (SOC misclassified as non-SOC). In addition, 34 calls, from 33 unique sequences, resulted in a false positive (FP) (non-SOC misclassified as a SOC).
We calculated aggregate metrics, taking the calls of all tools into account for a given sequence. For this, each tool was given a vote in classifying each sequence, and the majority vote for classification was compared with the NIST labels. This aggregate scoring scheme resulted in all sequences being correctly identified (i.e., at least four of six tools agreeing with NIST) and a majority of sequences (923, 92.4% of all sequences) were unanimously identified (i.e., 0 tools disagreed with the NIST label). Notably, there were only four sequences in total (0.4%) where as many as two tools disagreed with the NIST label of a sequence (Figure 1).
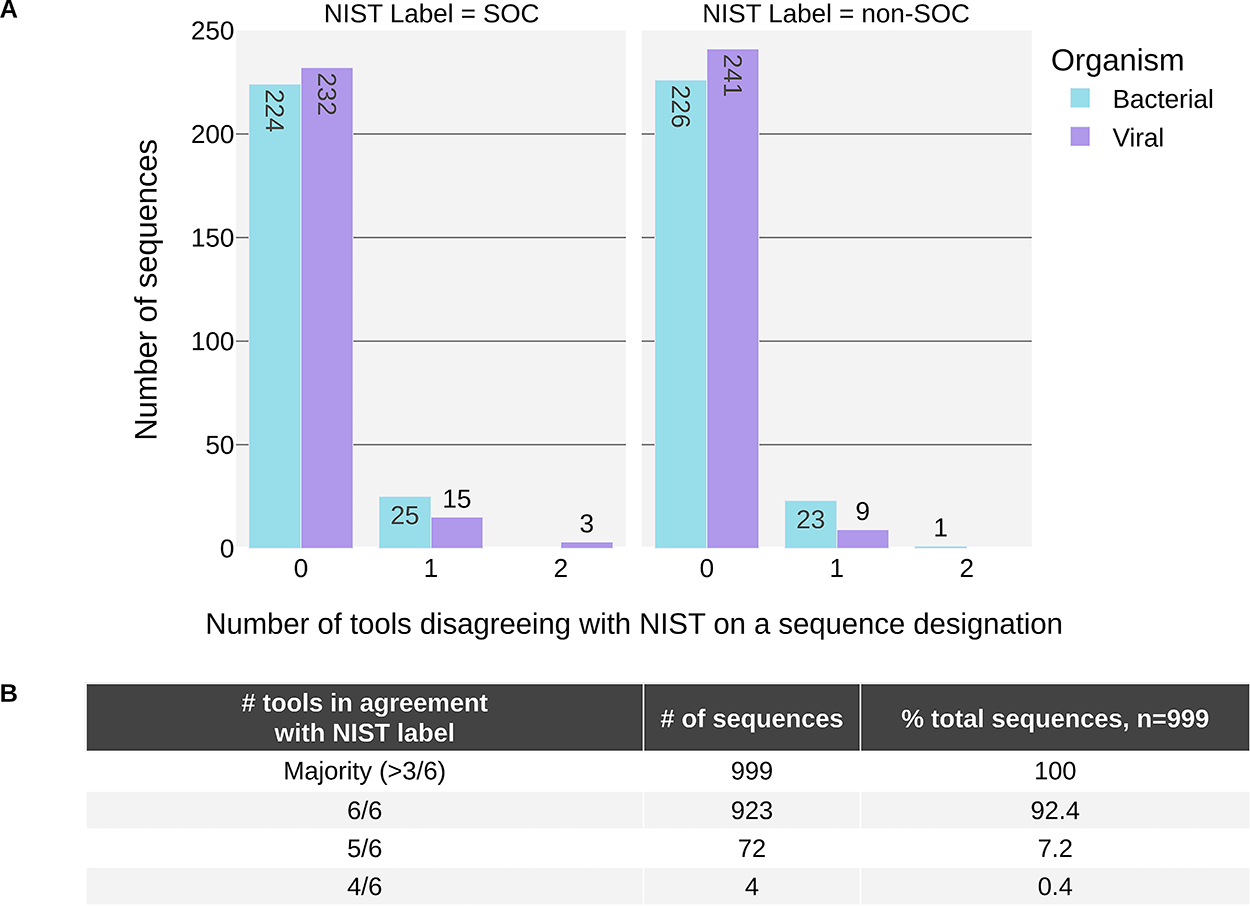
The majority of sequence classifications were in agreement with the NIST label.
When a tool disagreed with the NIST label for a sequence, it usually was alone, with the other five tools in agreement with NIST (72/76, 94.7% of disagreements), and most of these disagreements were bacterial sequences (49/72, 66.7% of disagreements) (Figure 1A). Notably, this is a taxonomic category where there is currently more uncertainty in the definition of risk: screening tools have been found to provide more “Undetermined” or “Optional” flagging determinations for bacterial sequences in contrast to viral sequences. 30
Screening tools effectively detect SOCs
Overall, the sequence screening algorithms performed well in classifying sequences in accordance with the NIST label. We calculated the false positive rate, false negative rate, accuracy, specificity, and sensitivity for all the tools. The sensitivity and accuracy obtained by all the tools were ≥95% and ≥97%, respectively (Table 3). We also calculated the negative predictive value (NPV) assuming a prevalence of 5% which represents a conservative estimate of the proportion of sequences flagged as SOCs in typical provider order streams. Notably, based on the measured sensitivities and specificities, the NPV of these tools is very high in low-prevalence settings.
Performance metrics of anonymized screening tools in random order
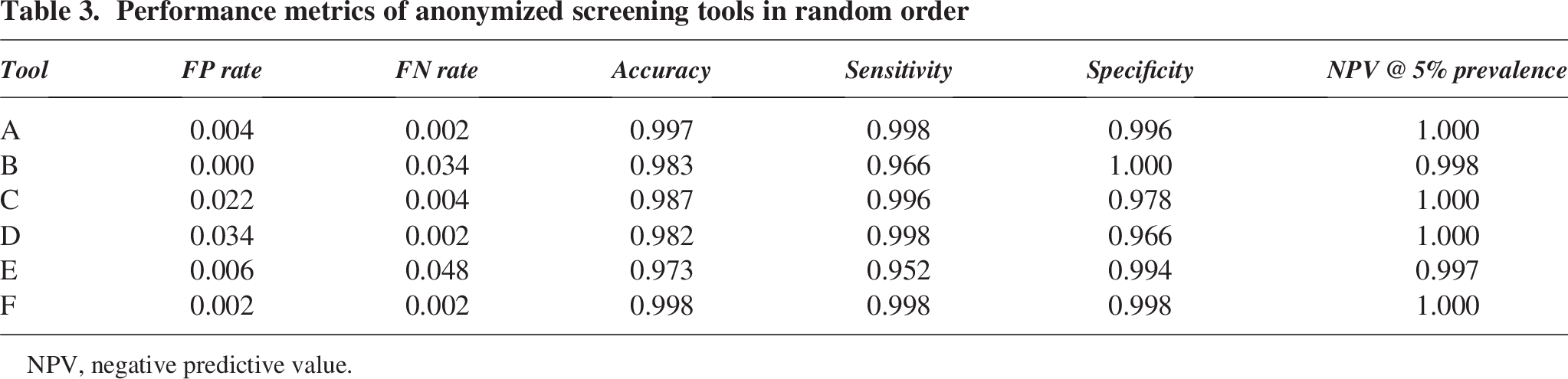
NPV, negative predictive value.
Disagreed-upon sequences are attributable to varying interpretations of the HHS guidance
Despite agreeing with a vast majority of the NIST labels, individual screening tools differed in their disagreements with labels (Figure 2). This is evident in Figure 2, which shows that disagreed-upon sequences (indicated in red) are for the most part unique to an individual tool (i.e., other rows in the column are for the most part gray).
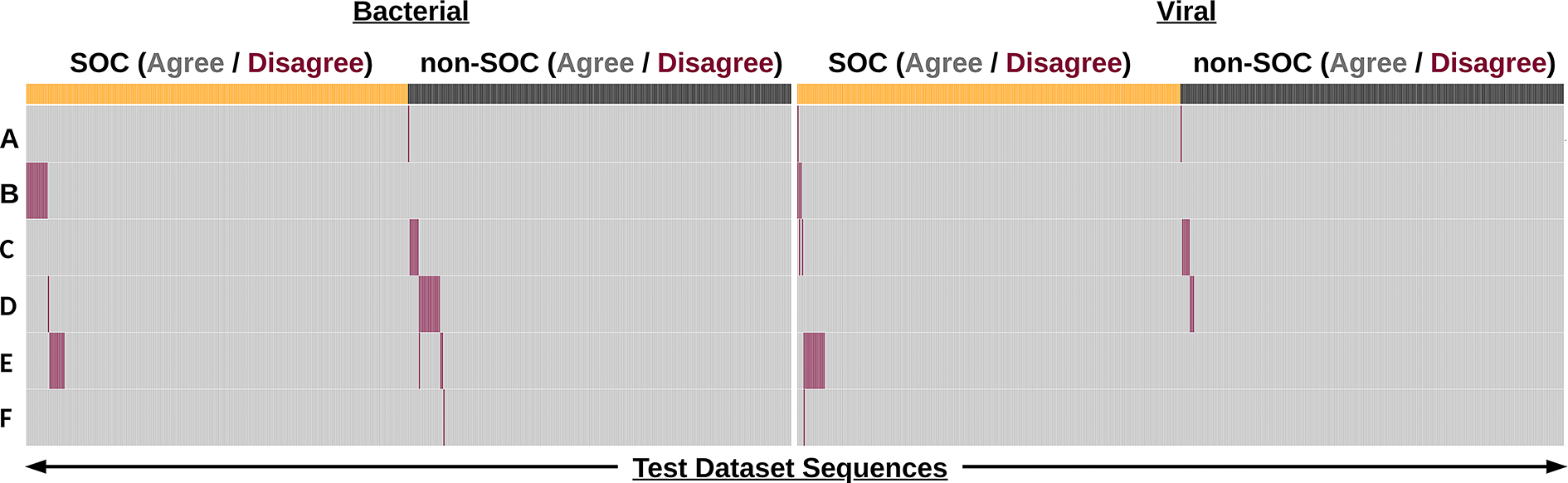
Sequence screening tools perform well on the NIST test dataset. Binary heatmap of sequence classification by anonymized tools
For sequence screening, avoiding FNs is the main priority. These represent potentially concerning sequence orders that are missed and not subjected to follow-up screening. While avoiding FNs is important, preventing FP sequences is also key to maintaining biosecurity. While not a direct hazard, FPs can lead to additional expenses in the form of subsequent customer screening/follow-up, as well as increasing screener fatigue. These expenses could, in turn, lead to an indirect biosecurity risk in the sense that TP sequences may be missed due to the increased screening burden. For the most part, the disagreements with the NIST labels (FPs/FNs) could be stratified into several categories based on differences in defining a SOC and/or methodological differences in screening algorithms. These definitional differences can be attributed to the characteristics of the sequence (NA vs. amino acid), debated sequence function (e.g., housekeeping vs. virulence), database curation (e.g., what are the sequences screened against for SOC determination and taxonomic assignment), and screening thresholds (e.g., what length of a match or how closely related does a match have to be for flagging a sequence). These definitional differences provide clarity as to why particular tools may disagree on a sequence and may be resolved by ongoing standardization activities. 30
NA uniqueness versus amino acid uniqueness (false negatives)
Two sequences (#910 and #984) were disagreed upon by two tools because they had an exact match to a nonregulated organism at the amino acid level, despite being unique to regulated organisms at the NA level. Sequence #910 is a 200-nt fragment of the dual specificity protein phosphatase from Mpox virus (Uniprot_acc: A0A7H0DN78, nucleotide region 1:201). While it is a unique match to Mpox genomes at the nucleotide level, it has exact matches at the amino acid level to other nonregulated members of Orthopoxvirus genus, such as Cowpox virus and Ectromelia virus. Sequence #984 is a 200-nt segment of the Genome polyprotein of Swine vesicular disease virus (Uniprot_acc: A0A8F5VSD2, nucleotide region 5001:5201) which, as far as our similarity searches indicate, is an exact match at the nucleotide level to only Swine vesicular disease virus genomes. However, at the amino acid level, it is an exact match to many other nonregulated genomes of the Enterovirus genus. While the HHS guidance states that an exact match to an unregulated organism is not a SOC, there is no specification on whether that is at the NA or amino acid level of a sequence. Nevertheless, the Department of Commerce has generally held uniqueness in amino acid space to be the dividing line.
Definition of a virulence factor (false negatives)
While NIST curated sequences that had been annotated as a “virulence factor” (based on experimental or inferred evidence), in some areas this annotation can be ambiguous or debatable. For instance, one tool disagreed with the label of sequence #259, which was a 200-nt component of the Carbamoyl phosphate synthetase gene (carB) of Francisella tularensis (UniProt_acc: Q5NEH1, nucleotide region 1001:1201) based on identifying it as matching a non-SOC “housekeeping” gene. For the purposes of sequence screening, “housekeeping” genes such as ribosomal RNAs (rRNAs), transfer RNAs (tRNAs), and metabolic genes have generally been exempted from being flagged due to their lack of relationship to pathogenicity or toxicity. No specific definition of “housekeeping” gene has ever been provided in screening guidance, however, and so it is unsurprising to find disagreements in this area.
Likewise, the notion of a virulence factor has multiple definitions, notably including the distinction between genes that directly implement virulence functionality (e.g., toxins, invasion proteins) versus genes associated with decreased virulence when disabled, which cover a much broader range of regulatory and supporting functionality. For example, the VFDB, from which the test sequences originated, lists carB as a “Nutritional/Metabolic” factor involved in virulence. While mutagenesis studies do implicate carB in counteracting reactive oxygen species and escaping the host phagosome,31,32 homologs of carB are found widespread in bacteria, where they play an essential role in metabolism and are not associated with virulence.
While NIST gathered sequences from BSAT genomes in the VFDB, there are known shortcomings with the VFDB when trying to identify SOCs based on function. 33 As the scope and definition of SOCs transition to include functional properties, the sequences included in standardized test datasets will require further scrutiny to determine if a sequence annotated as a virulence factor is indeed a sequence of concern. Identifying essential or important genes based on mutagenesis studies does not necessarily prove that a gene is involved in pathogenesis and thus a SOC.
Database differences (false negatives)
There were some sequences that disagreed upon due to differences in the databases used for screening. The definition of a SOC, as defined currently by the HHS guidance, relies heavily on sequence similarity between regulated and nonregulated organisms. Identifying a sequence as an exact/best match may differ depending on what sequence database(s) a particular algorithm uses.
In constructing the test dataset, NIST used only complete genomes from RefSeq to identify exact matches to virulence factor sequences. However, each screening tool includes different genomes and/or proteomes in its search databases that extend beyond what was in the NIST subset pulled from RefSeq (such as the nt or nr Genbank databases, UniRef, or even proprietary databases that are not publicly available; see Table 1). For example, sequence #372 was disagreed upon by one tool due to exact matches to nonregulated Burkholderia species. This sequence was a 200-nt portion of bsaU, a type III secretion system protein from B. pseudomallei (VFDB_acc: VFG002481, nucleotide region 1:201), which had exact matches to other B. pseudomallei and B. mallei genomes (both of which are regulated), as well as unclassified Burkholderia sp. 136 (2017), sp. 129, sp. 117, and sp. 137 (which are not regulated). Despite being in RefSeq, these unclassified genomes are only assembled to the “Contig” level and thus were not included in the similarity search, which would have removed them from the test dataset.
Interestingly, however, labeling the above genomes as “unclassified Burkholderia” is not the only valid classification. The Genome Taxonomy Database, which classifies bacterial and archaeal genomes according to phylogenomic analysis of single-copy marker genes, 34 groups these “unclassified” genome assemblies (GCF_002900705.1, GCF_002900675.1, GCF_002900725.1, GCF_002900745.1) with B. mallei. Thus, differences may come about not only due to the sequences included/excluded from a tool’s database but also from different taxonomic classifications given to the sequences. Along with the already known issues, 35 this highlights yet another challenge in using taxonomic lists for sequence screening.
Database differences (false positives)
Most FPs were likely the result of flagging due to sequence similarity to sequences from regulated agents. Notably, the construction of the NIST test dataset did not involve any sequence similarity searches for what were defined as TNs. Since these sequences were obtained from genomes of nonregulated organisms, any similarity search would identify a nonregulated sequence as an exact match according to the HHS guidance, thus categorizing it as a non-SOC. Nevertheless, these genomes from nonregulated organisms do, in some cases, have phylogenetic neighbors that are regulated. Most notably, this is the case with Escherichia coli K12 in our dataset, which is closely related to regulated E. coli and Shigella species. In fact, the one non-SOC disagreed upon by 2/6 tools was sequence #582. This sequence was a genome fragment from E. coli K-12 belonging to the aconitase hydratase B gene (which encodes an enzyme for the conversion of citrate to isocitrate in the Krebs cycle). While the K-12 strain is a common, nonpathogenic organism used routinely in biological research, sequence #582 was presumably flagged by those tools due to its close homology to sequences from pathogenic strains of E. coli (STEC, EHEC, VTEC), which are CCL-regulated agents, 36 although at the genetic level only the Shiga toxin gene is controlled. Thus, no tools should be calling nontoxin encoding E. coli sequences such as this one. Among all the FP sequences, E. coli K-12 sequences were the most abundantly disagreed-upon non-SOC category, accounting for 17/33 (52% of all false positive calls).
This category also presents an example whereby consensus-building activities between tool developers resulted in a reduction in false positives (Figure 3). A prior version of one tool that predated participating in consensus-building exercises (“X” in Figure 3) was used to analyze the test dataset. Results on this tool version showed high sensitivity to detect SOCs but lower accuracy when accounting for both SOCs and non-SOCs, compared with current versions of the tools. However, after database harmonization, that tool (one of “A” through “F” in Figure 3) improved its performance on the NIST test dataset to obtain both sensitivity and accuracy values in line with all other evaluated tools (above 95% and 97% respectively).
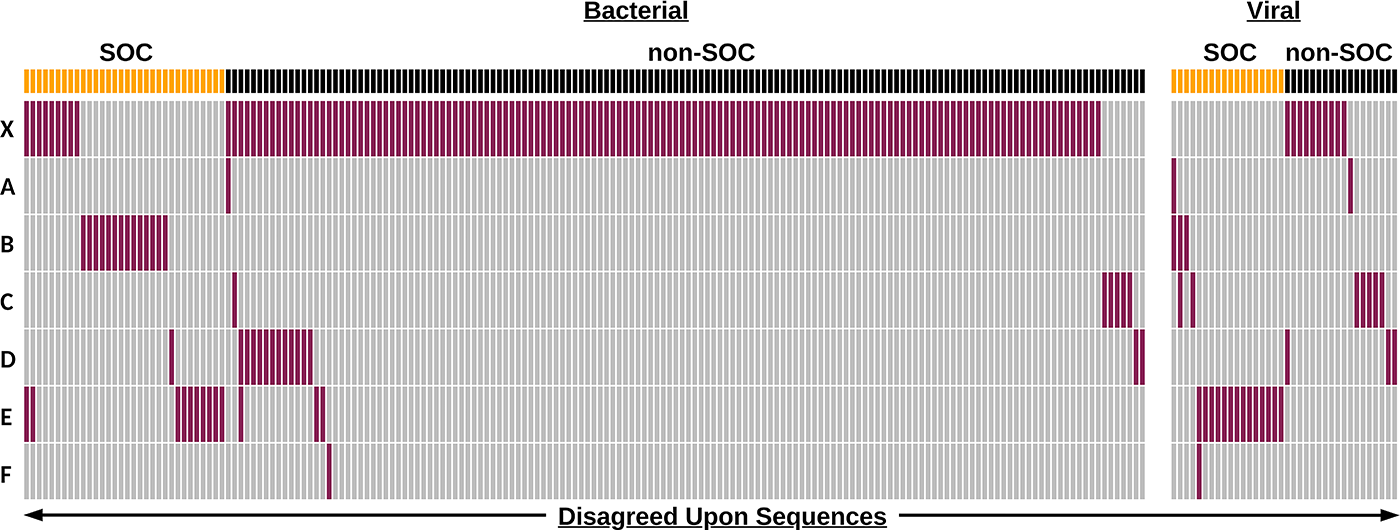
Disagreed-upon sequences are primarily bacterial and reduced through consensus building. Binary heatmap of sequence classification by anonymized tools. “X” represents an earlier version of one of the anonymized tools represented by “A” to “F.” Each column represents a sequence (total n = 214 including “X,” n = 76 for A–F only) that was misclassified. Sequences are partitioned into bacterial and viral categories and labels of SOC (yellow) and non-SOC (black) according to NIST labels. Sequence classifications are colored depending on if they agree (gray) or disagree (red) with the NIST label.
More stringent screening thresholds (false positives)
While the current definition of a SOC according to the HHS guidance relies on taxonomic similarity of sequences 200 nt or longer, the guidance recommends a shift toward defining SOCs based on function as soon as practicable, and by 2026 screening down to a minimum length of 50 nt. Notably, certain sequences were disagreed upon by tools because they are already screening in a manner that partly aligns with this more stringent definition. For example, one tool disagreed with the NIST label for sequence #683 (a genomic fragment of Enterobacteria phage T7) because a translated 63 nt segment of it contained a hit to the structural polyprotein of Chikungunya virus (a CCL agent).
The move toward a functional definition of “sequence of concern” is important since the current definition may omit concerning sequences from both controlled and noncontrolled agents. For instance, while constructing the test dataset, we identified certain segments of virulence factors with exact matches to nonregulated agents. There is ongoing work related to defining a sequence of concern based on functional properties.33,37–39 Notably, just because a sequence is an exact match to a nonregulated organism does not mean it is benign.
Implementation of the sequence screening assessments
Based on the results of the tool performance against the NIST test dataset, and in discussions with tool developers, key metrics were identified for a “passing score” when assessing baseline sequence screening. Since it is most critical to prevent a true SOC from being synthesized without verification of customer legitimacy, the main metric of concern is sensitivity. We identified a 95% (5% false negative rate) threshold for sensitivity and a 75% threshold for accuracy (see the Methods section) as currently achievable by all tested algorithms (Figure 3).
While most order streams typically contain a low frequency (<5%) of flagged sequences, we decided to have equal proportions of SOCs (TPs) and non-SOCs (TNs) in this test dataset in order to better assess the ability of algorithms to identify SOCs without having to evaluate an excessively large number of TN sequences. In order to assess baseline sequence screening, adequate power can be achieved with 200 TP and 200 TN sequences (see the Methods section), and additional sequences will be included that will go ungraded but serve as a way to obscure which sequences are being assessed, as well as providing a means for testing potential sequences to be graded in the future. To add more value and robustness to future test datasets, these ungraded sequences will include things that were not explicitly included in the initial test dataset, such as fungal-derived SOCs, an expanded variety of non-SOCs, and SOCs from non-BSAT and non-CCL pathogens. NIST plans to generate monthly test datasets and partner with international NGOs to host and administer blinded versions of these datasets to synthesis providers.
Conclusion
Taking into account the majority classification of sequences by current sequence screening algorithms, the NIST constructed test dataset captures the current definition of a SOC according to the available Guidance, which (prior to 2026) is focused on screening for best matches to federally regulated organisms on the BSAT list and/or CCL. While there were some differences in labeling SOCs and non-SOCs, these primarily localized to individual tools and appear to be due to slight definitional differences in what a tool developer or NIST identified as a SOC based on their respective interpretations of the HHS guidance. Ongoing interactions between tool developers and shared test datasets like the one described here provide a mechanism for harmonizing these interpretations. Notably, all of the tested tools currently screen sequences down to 50 nt and many of them implement screening against functional concerns, both of which are part of the guidance laid out for 2026. However, future work can still be done to further harmonize differences in SOC definitions among various tools and work toward a fully comprehensive functional definition of SOCs.
Our analysis of baseline performance suggests that many built-for-purpose synthesis screening tools are sufficiently adept to fit into robust NA synthesis screening workflows. Importantly, our analysis of tool performance enabled the identification of sensitivity and accuracy thresholds for test implementation. While this study was done with tool developers, the actual assessment will need to involve synthetic NA providers. While multiple providers may utilize the same tool, each tool can be run with different parameters (e.g., depending on the user or geographical location), and thus may lead to different screening outputs. In addition, providers may implement manual follow-up screening on certain sequences, thus further improving performance, especially in light of nuanced scenarios that led to FPs and FNs in this analysis. Therefore, NIST test datasets will be made available for use by synthetic NA providers to assess baseline sequence screening.
Footnotes
Acknowledgment
The authors thank Becky Mackelprang for providing valuable feedback on this article.
Funding Information
Work by K.F., C.B., B.G., J. Beal, T.M., S.T.M., T.A., and N.W. was partially supported by funding from Sentinel Bio. R.D. is supported in part by funds from the National Institutes of Health (P01-AI152999) and a training fellowship from the Gulf Coast Consortia, on the NLM Training Program in Biomedical Informatics & Data Science (T15LM007093). F.Q. is supported in part by funds from the National Institutes of Health (GCID U19 AI144297-05). M.N. is supported in part by funds from National Institutes of Health (P01-AI152999). T.J.T. is supported in part by funds from the National Science Foundation (IIS-2239114) and National Institutes of Health (P01-AI152999, GCID U19 AI144297-05).
Disclaimer
This document does not contain technology or technical data controlled under either U.S. International Traffic in Arms Regulation or U.S. Export Administration Regulations.
Certain equipment, instruments, software, or materials are identified in this article in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement of any product or service by NIST, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose.
Authors’ Contribution Statement
T.S.L.: Conceptualization, data curation, formal analysis, investigation, methodology, project administration, resources, software, validation, visualization, and writing—original draft. K.F.: Data curation, investigation, resources, software, and writing—review and editing. C.B.: Data curation, investigation, resources, software, and writing—review and editing. B.T.G.: Data curation, investigation, resources, software, and writing—review and editing. J. Beal: Data curation, investigation, resources, software, and writing—review and editing. T.M.: Data curation, investigation, resources, software, and writing—review and editing. S.T.M.: Data curation, investigation, resources, software, and writing—review and editing. J. Berlips: Data curation, investigation, resources, software, and writing—review and editing. L.F.: Data curation, investigation, resources, software, and writing—review and editing. R.D.: Data curation, investigation, resources, software, and writing—review and editing. F.Q.: Data curation, investigation, resources, software, and writing—review and editing. M.N.: Data curation, investigation, resources, software, and writing—review and editing. T.J.T.: Data curation, investigation, resources, software, and writing—review and editing. G.G.: Data curation, investigation, resources, software, and writing—review and editing. K.T.: Data curation, investigation, resources, software, and writing—review and editing. T.A.: Data curation, investigation, resources, software, and writing—review and editing. N.W.: Data curation, investigation, resources, software, and writing—review and editing. S.P.F.: Conceptualization, formal analysis, investigation, methodology, project administration, supervision, visualization, and writing—original draft.
Authors’ Disclosure Statement
K.F., C.B., B.G., J. Beal, T.M., S.T.M., J. Berlips, L.F., R.D., F.Q., M.N., T.J.T., G.G., K.T., T.A., and N.W. are affiliated with institutions that build and deploy biosecurity screening software.