
Abstract
A yeast artificial chromosome (YAC) containing a multigene cassette for expression of enzymes that enhance xylose utilization (xylose isomerase [XI] and xylulokinase [XKS]) was constructed and transformed into Saccharomyces cerevisiae to demonstrate feasibility as a stable protein expression system in yeast and to design an assembly process suitable for an automated platform. Expression of XI and XKS from the YAC was confirmed by Western blot and PCR analyses. The recombinant and wild-type strains showed similar growth on plates containing hexose sugars, but only recombinant grew on D-xylose and L-arabinose plates. In glucose fermentation, doubling time (4.6 h) and ethanol yield (0.44 g ethanol/g glucose) of recombinant were comparable to wild type (4.9 h and 0.44 g/g). In whole-corn hydrolysate, ethanol yield (0.55 g ethanol/g [glucose + xylose]) and xylose utilization (38%) for recombinant were higher than for wild type (0.47 g/g and 12%). In hydrolysate from spent coffee grounds, yield was 0.46 g ethanol/g (glucose + xylose), and xylose utilization was 93% for recombinant. These results indicate introducing a YAC expressing XI and XKS enhanced xylose utilization without affecting integrity of the host strain, and the process provides a potential platform for automated synthesis of a YAC for expression of multiple optimized genes to improve yeast strains.
Keywords
Introduction
Worldwide concern about improving energy security and achieving sustainable biofuel production has accelerated the development of renewable energy sources such as lignocellulosic biomass. Economical biomass conversion into biofuels and bioproducts will require the efficient and rapid fermentation of the sugars present in cellulosic hydrolysates. Hydrolysis of cellulosic biomass yields predominantly glucose and xylose.1–3 Saccharomyces cerevisiae is currently the most widely employed microbial catalyst for fermentation of glucose in the biotechnology industry, but this yeast does not naturally ferment xylose, although a few natural Saccharomyces strains capable of using xylose have been identified. 4 Commercialization of fuel ethanol production from cellulosic biomass has focused on engineering the glucose-fermenting industrial yeast S. cerevisiae to use pentose sugars. Extensive research efforts using innovative genetic engineering approaches have improved xylose utilization by S. cerevisiae,1,3,5–20 but no strain, either natural or engineered, has yet been reported to ferment xylose as efficiently as glucose.
S. cerevisiae naturally metabolizes xylulose; therefore, one approach to improving xylose utilization involves introducing the gene encoding xylose isomerase (XI), which catalyzes the direct conversion of xylose to xylulose and may be obtained from a microorganism naturally capable of fermenting xylose. Overexpression of endogenous xylulokinase (XKS), which catalyzes the conversion of xylulose to xylulose-5-phosphate, is also necessary to overcome the naturally low expression level of this enzyme,19,21 although very high levels have been shown to have an inhibitory effect during xylose fermentation, 22 suggesting levels of introduced enzyme activity must be consistent with the capacity of the surrounding metabolic network.
In previous work by Hughes and coworkers, 11 a three-plasmid yeast expression system using the portable small ubiquitin-like modifier (SUMO) vector set consisting of three vectors each with a different selectable marker (URA3, TRP1, or LEU2) was used to provide high expression levels of three proteins simultaneously to improve xylose utilization. In that study, a PCR assembly strategy was used to clone the Piromyces species E2 XI gene into the URA-selectable SUMO vector. The Yersinia pestis XKS gene was cloned into the LEU-selectable SUMO vector. A library of mutagenized genes encoding a cell-penetrating peptide was cloned into the TRP-selectable SUMO vector. The S. cerevisiae yeast strain INVSc1 was transformed with either all three SUMO plasmids or the SUMO-XI plasmid alone. Recombinant yeast strains expressing the three proteins showed improved aerobic growth rates in xylose liquid medium compared with the recombinant yeast expressing XI alone. 11
The SUMO system has been shown to increase expression levels of many different proteins and provides distinct advantages in gene fusion systems because (1) it increases solubility and stability of the recombinant proteins; (2) a highly specific, stable, and active SUMO protease is available in yeast for efficient fusion tag cleavage and subsequent purification of the recombinant protein; (3) native N-termini of recombinant proteins are preserved following SUMO protease cleavage; and (4) SUMO protease recognizes the tertiary SUMO structure with high specificity, thus never cleaving within the protein of interest. The enhanced expression and solubility of proteins or peptides fused to SUMO, combined with the broad specificity and highly efficient cleavage properties of SUMO protease, make this a useful system for obtaining high levels of expression of functional proteins in yeast.11,23,24
To demonstrate feasibility of a yeast artificial chromosome (YAC) as a stable protein expression system in S. cerevisiae, a synthetic chromosome was constructed containing a multigene cassette for expression of enzymes to enhance xylose utilization when stably transformed into S. cerevisiae. The multigene cassette contains the Piromyces species E2 XI gene with a SUMO yeast expression tag and the Y. pestis XKS gene obtained by PCR amplification from the plasmids used in our previous work 11 behind the TRP1 promoter. The pYAC4 plasmid was digested with restriction enzymes, and the SUMO-XI-XKS construct was ligated to the resulting YAC4 fragments (arms) to produce the artificial chromosome, YAC4-SUMO-XI-XKS. The pYAC4 plasmid was chosen because of its ability to incorporate large DNA inserts.25,26 It contains a yeast autonomously replicating sequence (ARS1) necessary for replication, with its associated centromere (CEN4) DNA sequence for segregation at cell division and two telomere-like DNA sequences (TEL) derived from Tetrahymena thermophila. 27 The centromere and telomere sequences allow maintenance of large cloned DNA as stable, single-copy yeast linear chromosomes.28,29 The S. cerevisiae yeast strain INVSc1 was transformed with YAC4-SUMO-XI-XKS, and the recombinant strain was evaluated for incorporation of the artificial chromosome and for xylose utilization.
The YAC expression system has advantages over plasmid-based and viral gene delivery systems for introducing genes of interest into cells because these systems are limited in the size of DNA sequences they can deliver. For applications where delivery of multiple gene open reading frames (ORFs) are desired, plasmid or viral vectors may be inadequate. In addition, they require integration into host chromosomes for stable maintenance. Such integrations are most often random, and the sites of integrations may have significant and unpredictable effects on the expression of introduced genes or may result in inactivation or change in regulation of host genes. Technologies that can accommodate large DNA inserts and do not require integration into the host genome for long-term stable maintenance would be advantageous, both in having more predictable gene expression and noninterference in host cell functions. 30
The YAC assembly and transformation processes were designed for use on an automated platform. The automated pipeline to assemble, isolate, and use the target YACs will be deployed as task-specific modules. Each module will consist of nodes; each node will satisfy work requests at the sample level. The system is intended to provide a potential synthetic biology platform for one-step construction of a yeast artificial chromosome containing a cassette for expression of multiple genes to further improve industrial yeast strains. 31
Materials and Methods
Assembly of YAC4-SUMO-XI-XKS Artificial Chromosome
The Piromyces species E2 XI gene ORF (GenBank accession no. AJ249909) was obtained by PCR amplification using Primers 1 and 2 (IDT, Coralville, IA; Table 1 ) with the plasmid pSUMOduo URA XI from Hughes et al. 11 as a template ( Fig. 1 , right top). PCR amplification was performed using a Phusion High-Fidelity PCR Kit (New England BioLabs, Ipswich, MA) according to the manufacturer’s instructions in a 50-µL volume. In addition, the AmpliTaq polymerase kit (Invitrogen, Grand Island, NY) was used to add adenine to the 5′ termini. The products were separated on an agarose (1% w/v) gel prepared in 300 mL of 1× TAE buffer (Sigma-Aldrich, St. Louis, MO) with 30 µL of 10 µg/mL ethidium bromide (Teknova, Hollister, CA) added. Gels were run in 1× TAE running buffer for 1.5 h at 100 V using a Bio-Rad Power Pac 3000 (Bio-Rad Laboratories, Hercules, CA). Images were taken with an AlphaImager 3400 (Alpha Innotech Corp., San Leandro, CA). The resulting SUMO-XI amplicon (1764 base pairs) was excised from the gel and purified using a GENECLEAN II PCR kit (MP Biomedicals, Solon, OH) according to the manufacturer’s directions. The amplicon was cloned into pCR8 (pCR8/GW/TOPO TA cloning kit; Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions to produce pCR8 SUMO-XI ( Fig. 1 , right center), and the plasmid was purified from TOP10 Escherichia coli using a QIAprep Spin Miniprep kit (QIAgen, Valencia, CA) according to the manufacturer’s instructions except that 100 µL EB buffer was used.
List of Oligonucleotide Primers Used to Construct Yeast Artificial Chromosome.
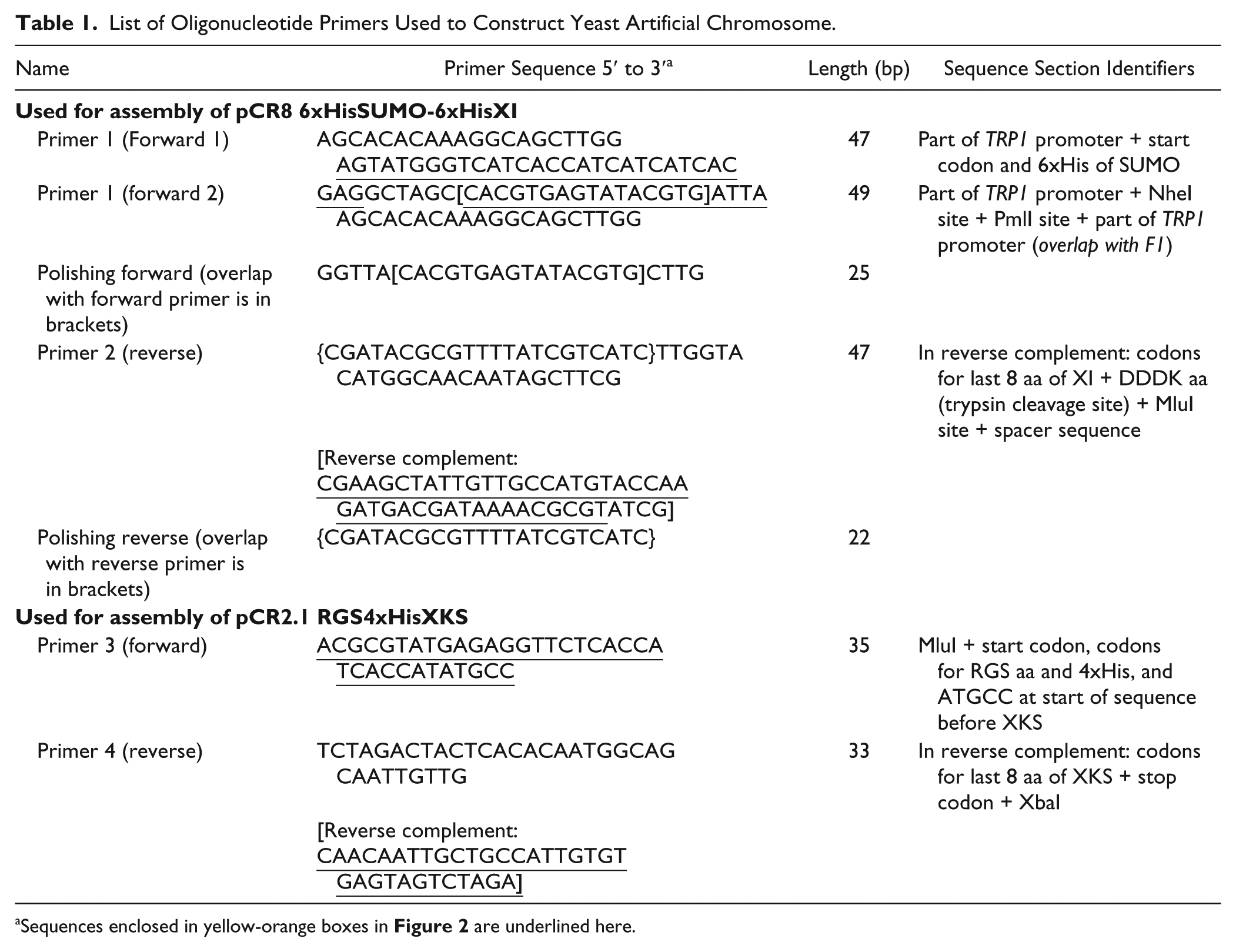
Sequences enclosed in yellow-orange boxes in Figure 2 are underlined here.
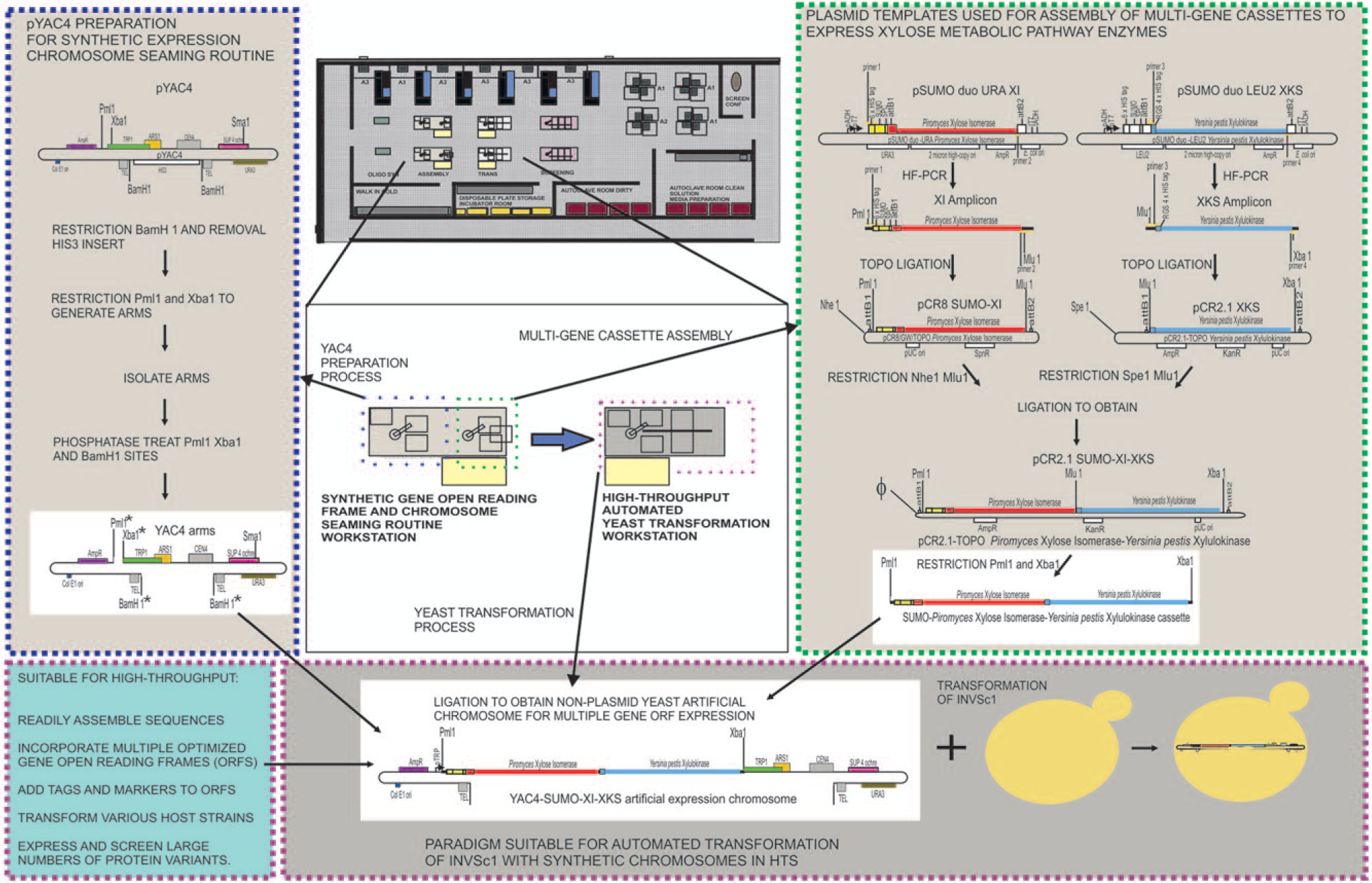
Assembly of a yeast artificial chromosome (YAC) containing a multigene cassette for expression of a polyprotein (SUMO-XI-XKS) and transformation into a host yeast. The two arms of the chromosome formed by restriction enzyme digestion of the pYAC4 plasmid (left) and the multigene cassette for expression of SUMO-XI-XKS formed by restriction of the plasmid pCR2.1 SUMO-XI-XKS (right) were combined to produce the artificial expression chromosome YAC4-SUMO-XI-XKS (bottom). Colors of sections correspond to colors of the nucleotide sequences in Figure 2 . Symbol (Φ) at left of pCR2.1 SUMO-XI-XKS represents one-half the SpeI site plus one-half the NheI site. Asterisks indicate dephosphorylated sites. SPC is the SUMO protease cleavage site. The YAC assembly and transformation processes are designed for implementation on the automated workstations pictured on the synthetic biology platform (center).
The Y. pestis XKS gene ORF (GenBank accession no. NP_671351) was obtained by PCR amplification using primers 3 and 4 ( Table 1 ) with the plasmid pSUMOduo LEU XKS from Hughes et al. 11 as a template ( Fig. 1 , right top). The resulting XKS amplicon (1530 base pairs) was cloned into pCR2.1 (pCR2.1 TOPO TA cloning kit; Invitrogen) after addition of 5′ adenine with AmpliTaq polymerase to produce pCR2.1 XKS ( Fig. 1 , right center), and the plasmid was purified from TOP10 E. coli using a QIAprep Spin Miniprep kit according to the manufacturer’s instructions except that 100 µL EB buffer was used.
The plasmid pCR8 SUMO-XI was digested with the restriction enzymes MluI and NheI (New England BioLabs), and the products were separated on an agarose (1% w/v) gel, from which the SUMO-XI fragment (1764 base pairs) was excised and purified using a GENECLEAN II PCR kit. The plasmid pCR2.1 XKS was digested with the restriction enzymes SpeI and MluI, and the products were separated on an agarose (1% w/v) gel, from which the pCR2.1 vector plus XKS fragment (5461 base pairs) was excised and purified using a GENECLEAN II PCR kit. The two purified DNA fragments were ligated to produce pCR2.1 SUMO-XI-XKS ( Fig. 1 , right bottom).
The plasmid pCR2.1 SUMO-XI-XKS was digested with the restriction enzymes XbaI and PmlI ( Fig. 1 , right bottom). At the same time, the plasmid pYAC4 was digested with the restriction enzymes BamHI, XbaI, and PmlI ( Fig. 1 , left), followed by addition of alkaline phosphatase (Antarctic Phosphatase; New England BioLabs) to dephosphorylate the 5′ DNA termini and prevent self-ligation of the two YAC4 fragments. The products of each of the two restriction digests were separated on agarose gels. The two arms from the pYAC4 digest (3.1 and 6.5 kb) and the SUMO-XI-XKS fragment from the pCR2.1 SUMO-XI-XKS digest (3294 base pairs) were purified separately. The three purified DNA fragments were combined (1:1:1 molar ratio) in a 20-µL ligation reaction (2 µL 10X T4 DNA ligase buffer, 1 µL T4 DNA ligase [20,000 U/µL; New England BioLabs], and 17 µL total of the purified DNA fragments) and incubated at 16 °C to produce the artificial expression chromosome YAC4-SUMO-XI-XKS ( Fig. 1 , bottom) that was transformed directly into S. cerevisiae.
Transformation of INVSc1 with YAC4-SUMO-XI-XKS
The S. cerevisiae yeast strain, INVSc1 (genotype: MATa his3Δ1 leu2 trp1-289 ura3-52/MATα his3Δ1 leu2 trp1-289 ura3-52), was obtained from Invitrogen. Transformation of the INVSc1 yeast was performed using an Alkali-Cation Yeast Transformation kit (MP Biomedicals) according to the manufacturer’s instructions with 2 mL yeast preculture grown overnight in 100 mL YPD medium to an absorbance at 660 nm equivalent to an optical density of 1.6 (DU-640 Spectrometer; Beckman Coulter, Fullerton, CA). Transformants were selected by plating onto 2% glucose complete minimal (CM) medium minus uracil plates (1.4 g yeast synthetic dropout medium supplement without histidine, tryptophan, leucine, or uracil [Sigma-Aldrich]; 6.7 g yeast nitrogen base with ammonium sulfate [Sigma-Aldrich]; 20 g D-glucose [Fisher Scientific, Fair Lawn, NJ]; 76 mg each of histidine, tryptophan, and leucine; and 20 g Bacto Agar [Sigma-Aldrich] in a final volume of 1 L Milli-Q water) or onto 2% xylose CM medium plates (1.4 g yeast synthetic dropout medium supplement without histidine, tryptophan, leucine, or uracil; 6.7 g yeast nitrogen base with ammonium sulfate; 20 g D-xylose; 76 mg each of histidine, tryptophan, leucine, and uracil; and 20 g Bacto Agar in a final volume of 1 L Milli-Q water).
PCR Analysis
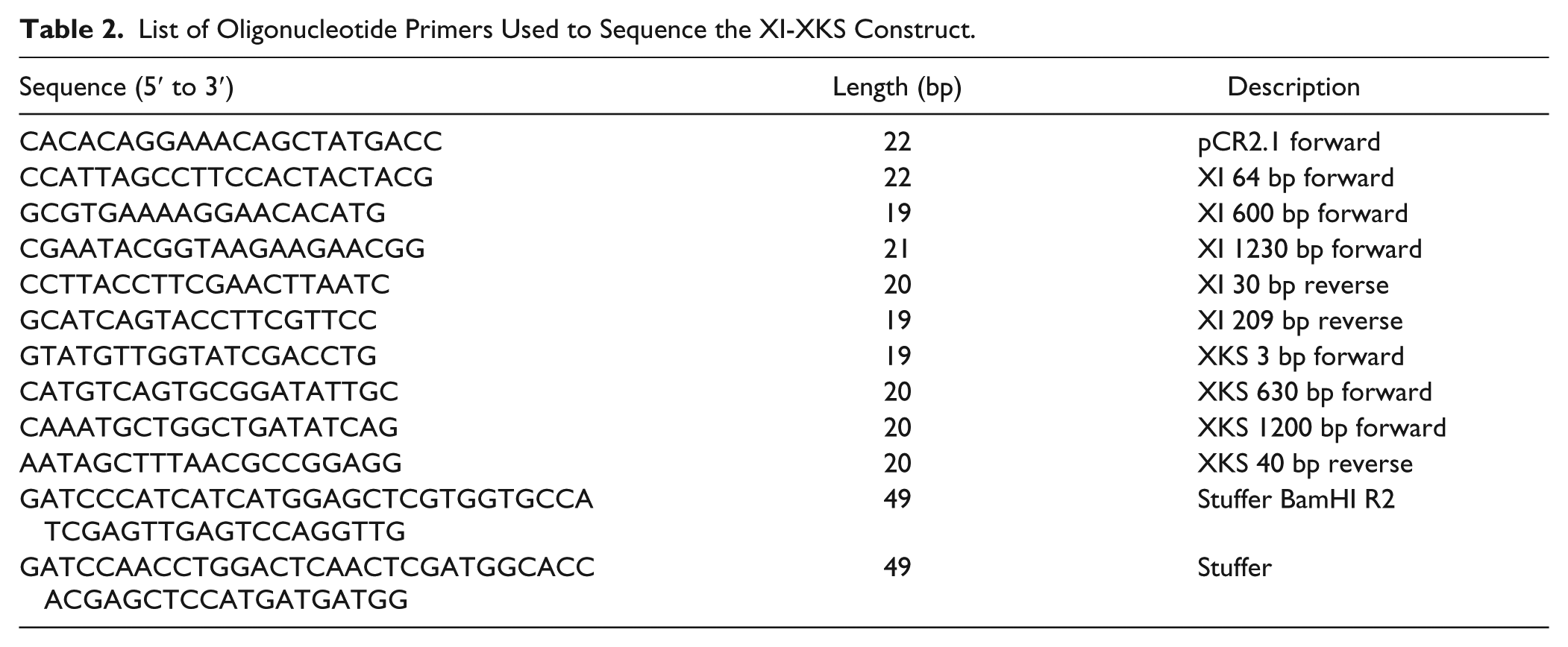
The sequence of the SUMO-XI-XKS insert was verified using the primers in Table 2 . Sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems by Life Technologies Corporation, Carlsbad, CA) according to the manufacturer’s protocols. The pCR2.1 SUMO-XI-XKS plasmid and a plasmid formed by ligating a stuffer fragment containing a SacII site for subsequent linearization into Klenow-treated BamHI sites of YAC4-SUMO-XI-XKS were used as templates.
List of Oligonucleotide Primers Used to Sequence the XI-XKS Construct.
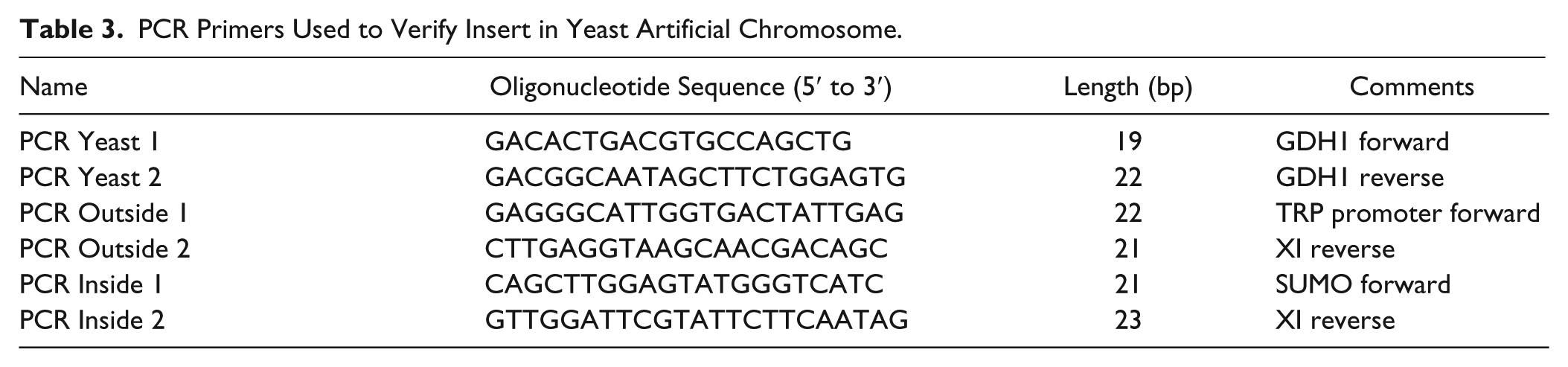
Genomic DNA was collected using the DNeasy Plant Kit (QIAgen) according to the manufacturer’s instructions. The PCR primers used to verify the presence of the artificial chromosome in the transformed INVSc1 yeast are given in Table 3 . The amplified DNA was analyzed by gel electrophoresis on agarose (1% w/v) gels stained with ethidium bromide. In total, 5 µL of a mix of HindIII digest of lambda DNA and HaeIII digest of phiX174 DNA (New England BioLabs) were loaded as size standards. Images were taken with an AlphaImager 3400.
PCR Primers Used to Verify Insert in Yeast Artificial Chromosome.
Western Blot and Coomassie Analyses
INVSc1 wild-type and INVSc1 YAC4-SUMO-XI-XKS strains were grown in 50-mL conical tubes containing 2% glucose CM medium (2% glucose CM minus uracil medium with 76 mg/L uracil added) or 2% glucose CM minus uracil medium, respectively, overnight at 30 °C at 100 rpm (New Brunswick Innova 4230 Shaker; Eppendorf, Enfield, CT) to an OD660 of 1.6, and the cells were pelleted at 3000 × g (Avanti J20 centrifuge with a 4.3 rotor; Beckman Coulter). The pellets were resuspended in 10 mL Y-PER (Sigma-Aldrich) containing protease inhibitors (Roche Molecular Biochemicals, Indianapolis, IN). After 1 h at room temperature, the cells were pelleted at 3000 × g, and the supernatant was decanted into a 50-mL tube that contained 0.1 mL Ni-NTA Superflow beads (QIAgen). The sample was incubated with tumbling at 4 °C for 2 h, and the beads were then pelleted by centrifugation at 3000 × g for 10 min. The supernatant was removed and the beads moved to a 1.5-mL microfuge tube. Then, 10 µL 0.5 M EDTA (Sigma-Aldrich) was added, and the mixture was vortexed. Next, 40 µL 2× Novex Tris-glycine loading buffer (Invitrogen) containing 2% β-mercaptoethanol (Sigma-Aldrich) was added, and the tubes were heated to 100 °C for 10 min. Then, 15 µL of each sample was loaded per well of the Novex sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) 4% to 20% gel (Invitrogen), and 10 µL SeaBlue Plus2 standards (Invitrogen) and 2.5 µL HIS-tagged protein ladder (QIAgen) were loaded in two of the wells. Gels were run in a Novex Xcell SureLock Mini-Cell (Invitrogen) at 125 V for 90 min in Novex 1× Tris-glycine running buffer (Invitrogen). Protein transfer to the PVDF membrane (Invitrogen) was performed in the Novex Mini-cell at 200 mA for 10 h in 1× Tris-glycine transfer buffer (Invitrogen). The membrane was developed using a Western Breeze Chromogenic (anti-mouse) Immunodetection Kit (Invitrogen) according to the manufacturer’s instructions. The primary antibody was anti–tetra His (QIAgen) at a concentration of 0.3 µg/mL. Western blot images were captured on an AlphaImager 3400 (Alpha Innotech Corp.).
Bands on a second SDS-PAGE gel were visualized by staining for 12 h in Coomassie blue (Bio-Rad). The gel was destained in 10% acetic acid and 20% methanol for about 6 h with four wash changes.
Spot Plate Analysis
Flasks of wild-type and recombinant strains were grown overnight at 30 °C on 20 mL CM glucose or CM glucose minus uracil medium, respectively, and the culture was diluted to an OD660 of 0.001. Then, 10 µL was spotted onto the plates with each of the media to be tested, and the plates were incubated at 30 °C for 3 days.
Scanning Electron Microscopy Analysis
Yeast cells grown in glucose medium, xylose medium, or corn hydrolysate were incubated aerobically at 30 °C for 12 h, suspended in saline (0.85% NaCl), and centrifuged to remove residual medium. The cells were then prepared for analysis following the procedure described previously. 11 The samples were subjected to scanning electron microscopy and analysis using a Zeiss Supra 40 Visible Pressure Field-Emission Scanning Electron Microscope at the Engineering and Mining Experiment Station, South Dakota School of Mines and Technology.
Fermentation Experiments
Fermentation experiments were carried out in 500-mL flasks with a working volume of 350 mL under semi-anaerobic conditions as described by Saha et al. 32 A liquid preculture was grown in a 100-mL flask for 2 days at 30 °C with shaking at 100 rpm (Innova 4230 Shaker; Eppendorf). The density of the preculture was adjusted to an OD660 of 0.1 (DU 800; Beckman Coulter), and 20 mL was added to 330 mL of the appropriate substrate (either 2.0% CM glucose with or without uracil or hydrolysate) with lactose added as an internal standard. One hydrolysate was prepared from ground whole corn (#2 yellow dent corn; Aventine Renewable Energy, Pekin, IL) by first extracting oil with hexane using a Soxhlet apparatus, 33 removing the hexane by reduced-pressure rotary evaporation (10 mbar, 30 °C) and then pretreating the defatted whole corn (15% solids, w/v) with dilute acid (0.5% H2SO4, v/v), and heating in a sealed bomb reactor at 120 °C for 1 h. The resulting slurry was adjusted to pH 5, then saccharified with three commercially available enzyme preparations—Celluclast 1.5L (Sigma-Aldrich), Novozyme 188 (Sigma-Aldrich), and Viscostar 150L (Dyadic Corp., Jupiter, FL)—followed by heating at 45 °C for 72 h and filter sterilized.34,35 The pH of the hydrolysate at inoculation was approximately 5. A second hydrolysate was prepared from finely milled spent coffee grounds using a 500-mL pretreatment reactor with 20% w/w solids in water, heating to 190 °C in a Techne Industrial fluidized sand bath (Bibby Scientific Limited, Staffordshire, UK), and holding for 15 min. The reactor was instantaneously decompressed/flashed to atmospheric pressure through a plug valve and the material collected. The steam-treated sample was adjusted to 10% w/w solids and treated with dilute H2SO4 for 1.5 h at 160 °C. The pH was adjusted to 4.8 to 5.0 with Ca(OH)2 and the sample treated with three commercially available enzyme preparations—Fiberzyme (Dyadic Corp.), Novozyme (Brentag North America, Reading PA), and Celluclast (Brentag)—followed by heating at 45 °C at 150 rpm for 72 h and filter sterilization. Experiments were conducted at 37 °C for 27 h in glucose, 120 h in whole-corn hydrolysate, and 96 h in hydrolysate from spent coffee grounds with agitation at 100 rpm. Samples were withdrawn periodically to determine cell growth (optical density observed at 660 nm) and concentrations of glucose, xylose, and ethanol.
Determination of Glucose, Xylose, and Ethanol Concentrations
Glucose, xylose, and ethanol concentrations were determined using a high-performance liquid chromatography (HPLC) separation system consisting of a solvent delivery system (P2000 pump; Spectra-Physics, San Jose, CA) equipped with an autosampler (717; Waters Chromatography Division, Millipore Corp., Milford, MA) and a computer software-based integration system (Chromquest 4.0; Spectra-Physics). An ion-moderated partition chromatography column (Aminex HPX 87H with Cation H micro-guard cartridge; Bio-Rad) was used. Samples (10 µL) were injected onto a heated column (65 °C) and eluted at a flow rate of 0.6 mL/min with 5 mM H2SO4. Because corn hydrolysate samples were not diluted prior to HPLC analysis, injection volume was 1 µL. Peaks were detected with a refractive index detector (410 differential refractometer; Waters Chromatography Division, Millipore Corp.) and were identified and quantified by comparison to retention times of authentic standards.32,36
Results and Discussion
Construction of Artificial Chromosome YAC4-SUMO-XI-XKS
The artificial chromosome YAC4-SUMO-XI-XKS constructed in this study was designed to express the SUMO-XI-XKS fusion protein behind the TRP1 promoter and was assembled as shown in Figure 1 using the oligonucleotide primers listed in Table 1 to amplify the SUMO-XI and XKS sequences from plasmids used in previous work. 11 A similar process to that used in this study could be followed to assemble a polyprotein expression cassette containing in-frame multiple optimized synthetic gene ORFs for expression of enzymes and other proteins to enhance xylose utilization1,3 or for insertion of gene ORFs expressing a valuable peptide product to improve cost-effectiveness of cellulosic biofuel production. 37 Yeast artificial chromosomes are ideal for multigene cassette insertion because they allow for stable incorporation of DNA fragments larger than 100 kb. 26
The assembly and transformation processes are designed for use in an automated format, and the automated pipeline to assemble, isolate, and use the target YACs will be deployed as task-specific modules. Each module will consist of nodes; each node will satisfy work requests at the sample level. The architecture of the pipeline will enable the addition and removal of nodes on-demand, dynamically altering the throughput of the pipeline as protocols are optimized and/or added/dropped from the pipeline. Examples of modules are Multi-Gene Cassette Assembly, YAC Preparation, Chromosome Assembly, Transformation, Plating, Colony Picking, Plasmid Preparation, Protein Expression, and Protein Purification. This system provides a potential synthetic biology platform ( Fig. 1 , center) for one-step construction of a YAC containing a cassette for expression of multiple genes and transformation of the YAC into yeast host strains.
The nucleic acid sequence of the XI-XKS gene construct in the artificial chromosome was verified using the oligonucleotide primers listed in Table 2 . The nucleic acid sequence for the entire artificial chromosome and the amino acid sequence for the SUMO-XI-XKS polyprotein are presented in Figure 2 . The colors of the nucleic acid sequences shown in Figure 2 correspond to the colors of those sequences in the diagram of the assembly process ( Fig. 1 ) and are TEL (gray), ori E. coli (dark blue), AmpR (plum), TRP1 promoter (before 6xHis-SUMO start codon), 6xHis-SUMO (yellow; final two triplets code for SUMO protease cleavage [SPC] site), 6xHis-XI (red; third from last triplet codes for trypsin cleavage site), RGS4xHis-XKS (blue; RGS sequence comes from the plasmid used as template for XKS), TRP1 (green), ARS1 (orange-brown dotted box), CEN4 (gray), SUP4 (pink), URA3 (yellow-brown), and TEL (gray). Sequences of the assembly primers—primer 1 (forward 1 + forward 2), primer 2 (reverse), primer 3 (forward), and primer 4 (reverse), listed in Table 1 —are enclosed in yellow-orange boxes in Figure 2 . The sequences important for maintaining large cloned DNA as a stable yeast linear chromosome are the yeast autonomously replicating sequence (ARS1) necessary for replication, the centromere (CEN4) DNA sequence for segregation at cell division, and the two telomere-like DNA sequences (TEL).27–29

Nucleic acid sequence of yeast artificial chromosome YAC4-SUMO-XI-XKS. The amino acid sequence for SUMO-XI-XKS is provided below the nucleic acid sequence coding for these proteins. The colors of the nucleic acid sequences correspond to the colors of those sequences in the diagram of the assembly process ( Fig. 1 ) and are TEL (gray), ori Escherichia coli (dark blue), AmpR (plum), TRP1 promoter (before 6xHis-SUMO start codon), 6xHis-SUMO (yellow; final two triplets code for SUMO protease cleavage site), 6xHis-XI (red; third from last triplet codes for trypsin cleavage site; black bar between XI and XKS on YAC in Fig. 1 ), RGS4xHis-XKS (blue), TRP1 (green), ARS1 (orange-brown dotted box), CEN4 (gray), SUP4 (pink), URA3 (yellow-brown), and TEL (gray). Assembly primers 1 to 4 listed in Table 1 are enclosed in yellow-orange boxes.
Expression of XI and XKS in INVSc1 Yeast Transformed with YAC4-SUMO-XI-XKS
Western blot and Coomassie analyses of purified proteins from INVSc1 yeast transformed with the yeast artificial chromosome YAC4-SUMO-XI-XKS compared with proteins from INVSc1 without the artificial chromosome are presented in Figure 3 . In the Western blot analysis, the bands are visualized by chromogenic immunodetection using the anti–tetra His antibody that binds to the 6xHis sequence preceding the SUMO tag, the 6xHis sequence before XI, and the 4xHis sequence before XKS. In the lane for INVSc1 transformed with YAC4-SUMO-XI-XKS, bands are observed at 55, 52, and 50 kD, which are not detectable in the lane for wild-type INVSc1 that does not contain the artificial chromosome. These bands correspond to the molecular weights expected for RGS4xHis-XKS, attB1-6xHis-XI (from SUMO protease cleavage), and 6xHis-XI (translated from ATG before the codons for 6xHis), respectively. SUMO protease cleavage (SPC) occurs at the SPC site (glycine-glycine seen at the SUMO terminus in Fig. 2 and labeled SPC in Fig. 1 ). The linkage between XI and XKS is cleaved at the trypsin cleavage site (DDDK following the XI sequence in Fig. 2 ) by a yeast trypsin protease. The 12-kD 6xHis-SUMO fragment is expected at 15 to 18 kD. 38 On the Coomassie gel, the three bands at the locations expected for XKS and XI proteins are observed with greater intensity. In addition, a faint band is seen on the Coomassie-stained gel above 98 kD, at approximately 119 kD, which would correspond to the molecular weight of the XI-XKS polyprotein.
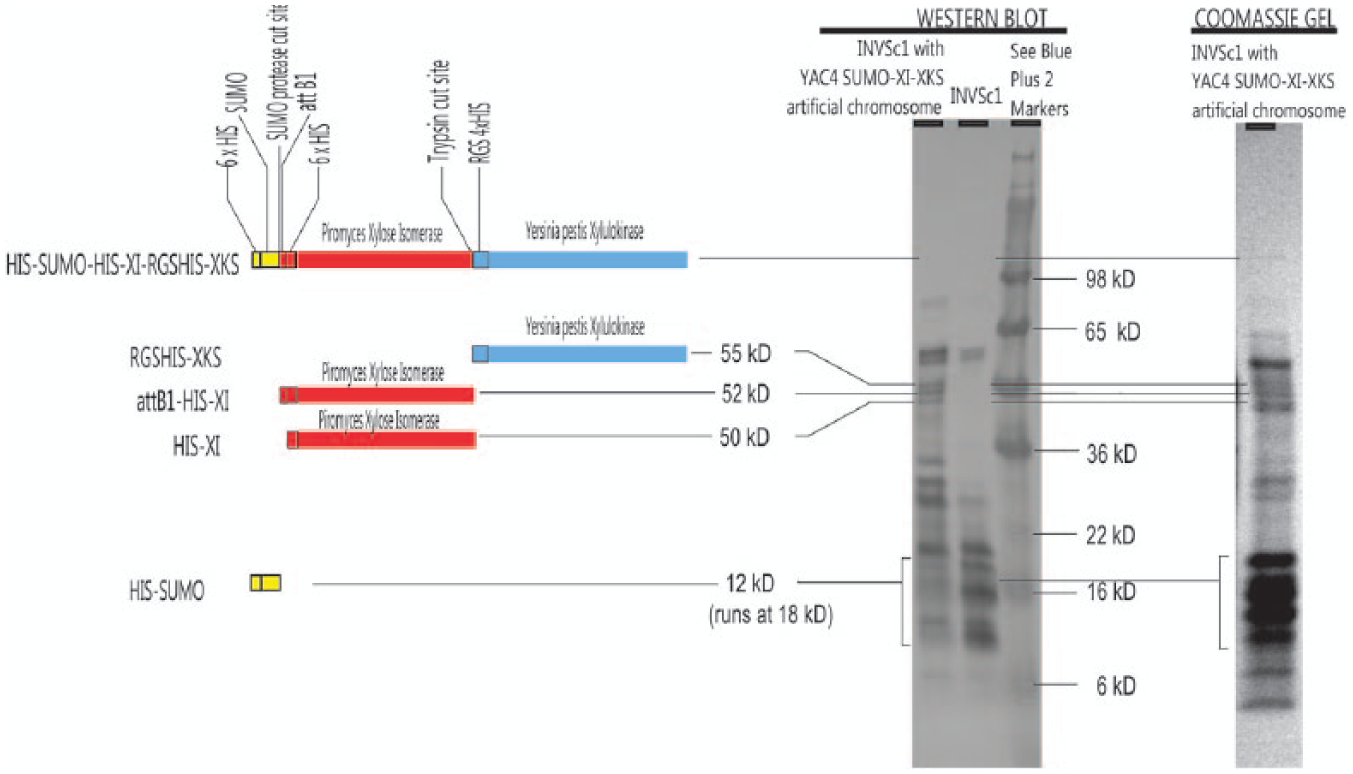
Western blot and Coomassie analyses of purified proteins from INVSc1 yeast transformed with the yeast artificial chromosome, YAC4-SUMO-XI-XKS, compared with proteins from wild-type INVSc1 not containing the artificial chromosome. In the lane for INVSc1 transformed with YAC4-SUMO-XI-XKS, bands are observed at 55, 52, and 50 kD, which are not detectable in the lane for INVSc1 without the artificial chromosome.
Fusion of a SUMO tag to the N-terminus of proteins has been found to enhance their expression and solubility. It provides distinct advantages in gene fusion systems11,23 because yeast cells contain a protease (Ulp1 or SUMO protease 1) that can cleave a variety of SUMO fusions. 24 Expression of both XI and XKS uses the TRP1 promoter. Expression of a polyprotein gene under the control of one promoter on an artificial chromosome transformed into the yeast cell would provide consistent and balanced expression levels not seen in plasmid-based systems. In previous work in our laboratory, 11 it was observed that introduction of an increasing number of plasmids decreased the amount of protein expressed by each plasmid.
PCR Amplification from YAC4-SUMO-XI-XKS in Recombinant Yeast
To demonstrate that the chromosomal DNA of the transformed yeast strain remained unchanged, a known genomic sequence unique to S. cerevisiae was identified and the genomic DNA of the INVSc1 transformed with YAC4-SUMO-XI-XKS was subjected to PCR amplification using primers to that sequence. The genomic sequence coding for S. cerevisiae glutamate dehydrogenase (GDH) on chromosome XV was selected, and a 623-bp section of the gene sequence was amplified in both the recombinant and wild-type strains and in a pYAC4 SUMO-XI-XKS plasmid (a closed version of the YAC; does not have HIS3 sequence) as a control (left-hand box of Fig. 4 ). The forward and reverse GDH1 PCR primers are listed in Table 3 . The PCR amplification products from genomic DNA of the INVSc1 strain transformed with YAC4-SUMO-XI-XKS compared with products from genomic DNA of wild-type INVSc1 separated on an agarose gel are shown in Figure 4 (left-hand box). The 623-bp amplicon from the S. cerevisiae GDH genomic sequence is present in both the recombinant strain and the wild-type strain, suggesting that the chromosomal DNA of INVSc1 is not altered by transformation with YAC4-SUMO-XI-XKS. The 623-bp amplicon was not observed in the lane for the control plasmid.
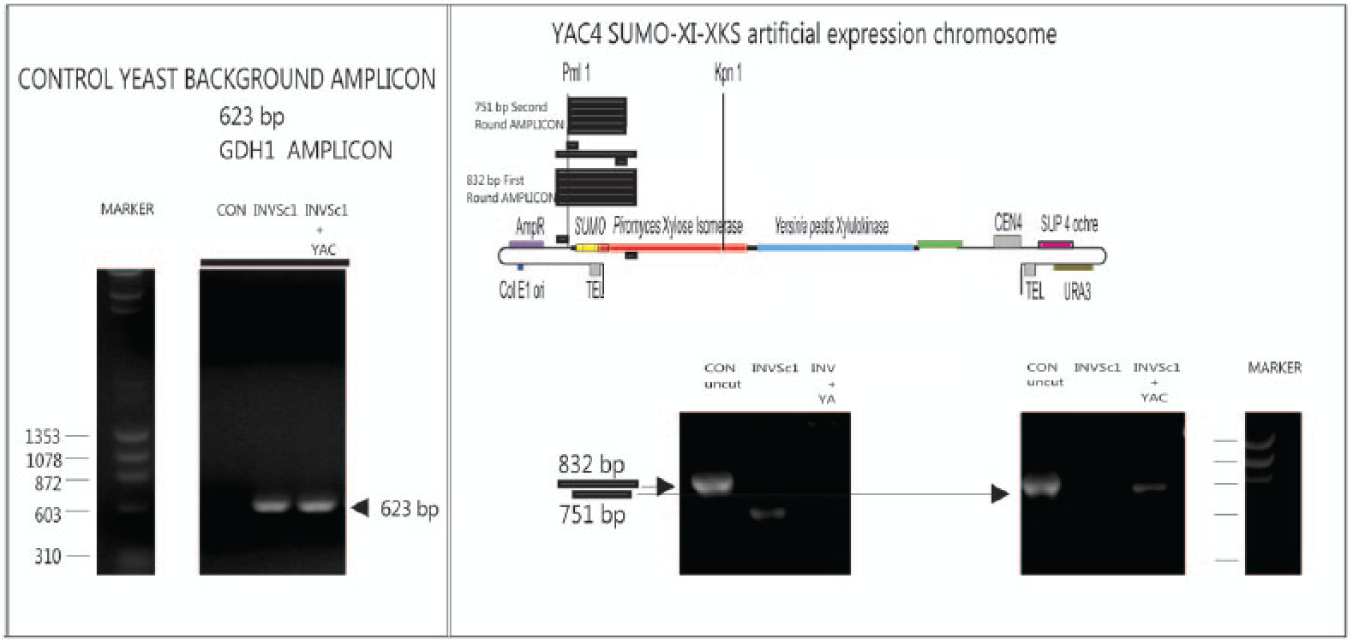
PCR amplification products from the artificial chromosome separated on an agarose gel. Left-hand box: A 623-bp section of genomic sequence coding for Saccharomyces cerevisiae glutamate dehydrogenase (GDH) on chromosome XV amplified in both the recombinant and wild-type strains and not in the control (pYAC4 SUMO-XI-XKS plasmid) demonstrates the native yeast genome is unchanged in the recombinant strain. Right-hand box: Two rounds of PCR using the pYAC4 SUMO-XI-XKS plasmid and the genomic DNA of both the INVSc1 strain transformed with YAC4-SUMO-XI-XKS and the wild-type INVSc1 strain. First-round PCR amplified an 815-bp region in the artificial chromosome and pYAC4 SUMO-XI-XKS plasmid control (left gel). The lane labeled as INVSc1 + YAC would be expected to contain the band for this amplicon, but it is too faint to detect. Therefore, a second PCR was performed to amplify a 753-bp region within the region amplified by the first PCR. The gel on right indicates the presence of the 753-bp amplicon in the recombinant strain and the pYAC4 SUMO-XI-XKS control plasmid and its absence in the wild-type strain.
To verify the presence of the YAC4-SUMO-XI-XKS in the recombinant strain, the pYAC4 SUMO-XI-XKS plasmid and the genomic DNA of both the INVSc1 strain transformed with YAC4-SUMO-XI-XKS and the wild-type INVSc1 strain were subjected to two rounds of PCR. The first round of PCR used primers that would amplify an 815-bp region in the artificial chromosome and the pYAC4 plasmid beginning in the region including the PmlI site and the TRP1 promoter and ending 385 nucleotides after the start codon of XI. The presence of the 815-bp amplicon identifies the insert corresponding to this region of the YAC-XI. The lane labeled CON plasmid on the gel pictured on the left-side of the right-hand box in Figure 4 indicates that this amplicon is present in the plasmid. The lane labeled as INVSc1 + YAC would be expected to contain this band amplicon, but it is too faint to detect. The band corresponding to this amplicon was absent in the lane labeled as INVSc1 (without YAC), although there is a contaminating band, so it is difficult to be certain. Therefore, to definitively determine that the region was present in the recombinant strain, a second amplification was performed on the reaction mixture using a set of primers that would amplify a 753-bp region within the region amplified by the first round of PCR with the first amplicon as the template. The gel pictured on the right side of the right-hand box in Figure 4 indicates that the 753-bp amplicon is present in the recombinant strain and that no 815-bp amplicon was present in the wild-type strain, verifying the presence of the YAC in the recombinant strain and its absence in the wild-type strain. Sequences of PCR primers used are given in Table 3 .
Growth on Plates of INVSc1 Yeast Transformed with YAC4-SUMO-XI-XKS
The growth of INVSc1 transformed with the yeast artificial chromosome expressing XI and XKS compared with wild-type INVSc1 on spot plates with YPD medium or CM media containing D-galactose, D-glucose, L-arabinose, D-mannose, D-xylose/agar, D-xylose minus uracil, D-glucose minus uracil, or D-xylose/agarose is shown in Figure 5 . Both strains grew very well on galactose, and the amount of growth was essentially the same for both. Both strains grew almost as well on glucose and mannose, with the recombinant strain demonstrating slightly better growth. Growth of both strains was best on YPD because of the addition of protein and yeast cell hydrolysates, with the recombinant strain demonstrating slightly better growth. Growth of the recombinant strain was observed on xylose and arabinose but much less than on the hexose sugars. Growth was slightly better on xylose than arabinose. Essentially, no growth was observed on arabinose with the wild-type strain. A very faint spot was seen on xylose/agar with the wild-type strain. When xylose/agarose was used instead of xylose/agar, essentially no growth was observed for the wild-type strain while growth was observed for the recombinant strain. The wild-type strain showed no growth on glucose medium minus uracil or xylose medium minus uracil, while the growth of the recombinant strain on glucose medium minus uracil or xylose medium minus uracil was similar to its growth on either medium with uracil.

Growth of INVSc1 YAC4-SUMO-XI-XKS compared with wild-type INVSc1 on spot plates. In total, 10 µL of cultures of wild-type and recombinant strains diluted to an OD660 of 0.001 was spotted onto plates with each of the media indicated, and the plates were incubated at 30 °C for 3 days.
Scanning Electron Micrographs of INVSc1 YAC4-SUMO-XI-XKS Compared with Wild-Type INVSc1
Scanning electron micrographs of the INVSc1 YAC4-SUMO-XI-XKS strain compared with the wild-type INVSc1 strain are shown in Figure 6 . The general shape of the cells and pattern of growth are similar for both the recombinant strain and the wild-type strain when grown on glucose medium and when grown on whole-corn acid hydrolysate. However, when grown on xylose, the cells of the recombinant strain appear more viable than the cells of the wild-type strain; the growth of the cells of the wild-type strain on xylose is less vigorous. The growth of the recombinant strain on xylose is similar to its growth on glucose, suggesting that the recombinant strain is able to use xylose for growth.
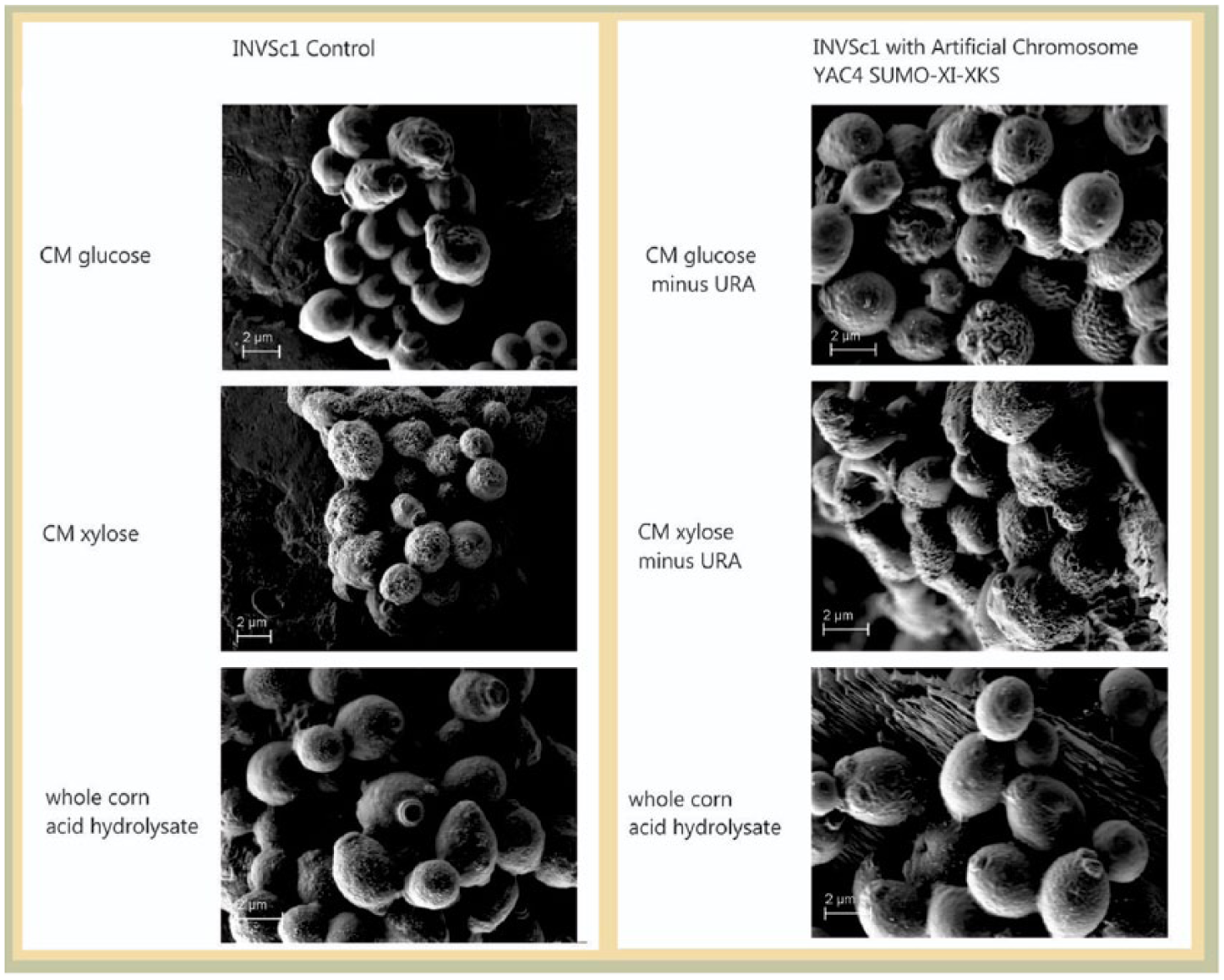
Scanning electron micrographs of INVSc1 YAC4-XI-XKS compared with wild-type INVSc1 grown in glucose medium, xylose medium, or whole-corn hydrolysate.
Fermentation Experiments Comparing Growth, Substrate Consumption, and Ethanol Production of INVSc1 YAC4-SUMO-XI-XKS and Wild-Type INVSc1
Growth, substrate consumption, and ethanol production during fermentation experiments with glucose medium or whole-corn acid hydrolysate comparing the wild-type strain with the recombinant strain are depicted in Figure 7 and summarized in Table 4 . With glucose as the sole carbon source, substrate consumption (100% at 21 h) and maximum ethanol yield (0.44 g ethanol/g glucose at 21 h) were the same for both strains ( Fig. 7A ). Doubling times for the wild-type and recombinant strains, 4.9 h and 4.6 h, respectively ( Table 4 ), were comparable. These results indicate that introduction of the yeast artificial chromosome did not alter the glucose utilization characteristics of the wild-type yeast strain.
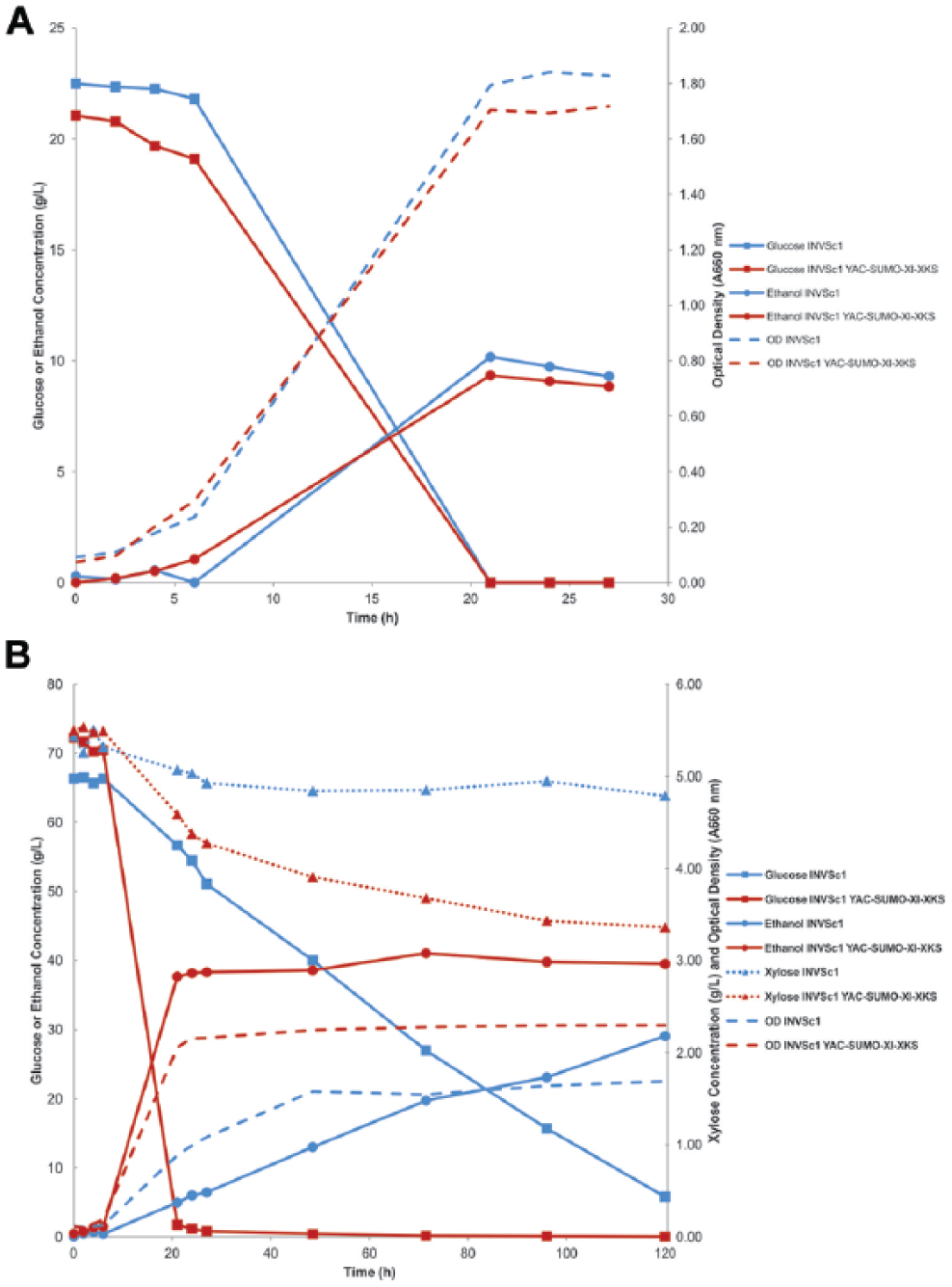
Growth, substrate consumption, and ethanol production of INVSc1 YAC4-SUMO-XI-XKS compared with wild-type INVSc1 during fermentation experiments. (
Comparison of Growth, Substrate Consumption, and Ethanol Production of Wild-Type INVSc1 vs. INVSc1 YAC-SUMO-XI-XKS in Fermentation Experiments with Glucose or Whole-Corn Hydrolysate.
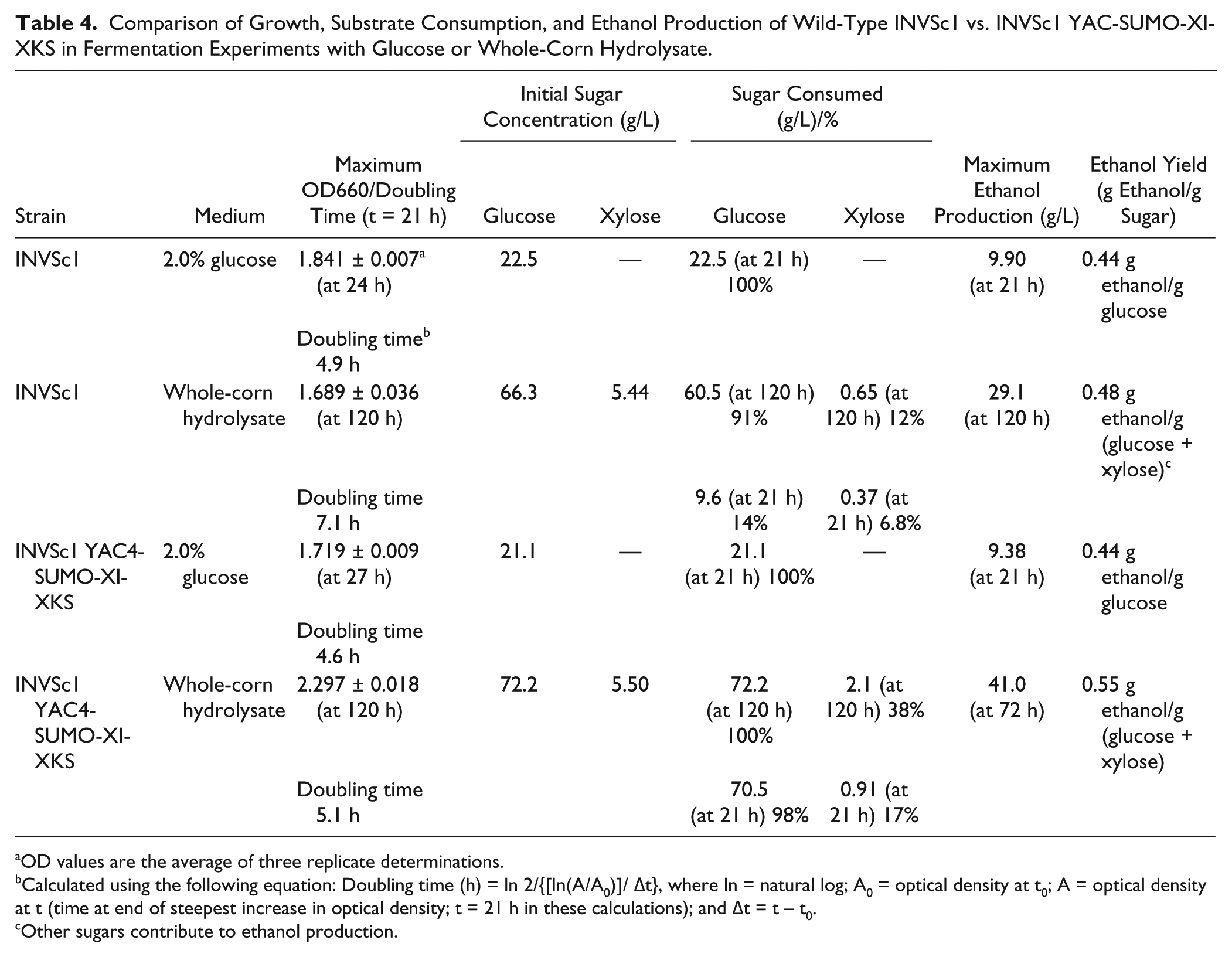
OD values are the average of three replicate determinations.
Calculated using the following equation: Doubling time (h) = ln 2/{[ln(A/A0)]/ Δt}, where ln = natural log; A0 = optical density at t0; A = optical density at t (time at end of steepest increase in optical density; t = 21 h in these calculations); and Δt = t – t0.
Other sugars contribute to ethanol production.
When whole-corn acid hydrolysate was the carbon source ( Fig. 7B ), the recombinant strain maintained a doubling time (5.1 h) similar to that when grown on glucose (4.6 h). However, with the whole-corn acid hydrolysate, the doubling time (7.1 h) of the wild-type strain was approximately 1.5 times greater than it was with glucose (4.9 h) as the sole carbon source ( Table 4 ). Glucose in the hydrolysate was 98% consumed at 21 h with the recombinant strain, but only 14% of the glucose was consumed with the wild-type strain at 21 h, and at 120 h, 91% was consumed. At 120 h, xylose consumption in the whole-corn hydrolysate was 38% of the initial xylose for the recombinant strain and 12% for the wild-type strain. Maximum ethanol production from whole-corn hydrolysate for the recombinant strain was 41.0 g/L. Based on the amounts of glucose and xylose consumed, the ethanol yield was 0.55 g ethanol/g (glucose + xylose) for the recombinant strain. Maximum ethanol production from whole-corn hydrolysate for the wild-type strain was 29.1 g/L. Based on the amounts of glucose and xylose consumed, the ethanol yield was 0.48 g ethanol/g (glucose + xylose) for the wild-type strain. Other sugars contribute to ethanol production, but glucose and xylose are the major sugar constituents of the hydrolysate.
When a hydrolysate of spent coffee grounds was the carbon source ( Fig. 8 ), the doubling time of the recombinant strain (4.3 h) was similar to that when grown on glucose as the carbon source (4.6 h). The recombinant strain consumed 91% of the initial amount of glucose (11.0 g/L) and 93% of the initial amount of xylose (13.3 g/L) in 20 h. Maximum ethanol production was 10.1 g/L. Based on the amounts of glucose and xylose consumed, the ethanol yield was 0.46 g ethanol/g (glucose + xylose) for the recombinant strain. The recombinant yeast strain has potential application for utilization of the cellulosic and oil portions of corn in temperate regions such as the United States, where corn is grown, or for the complex mixture of materials in spent coffee grounds in tropical countries such as Colombia, where coffee is grown.
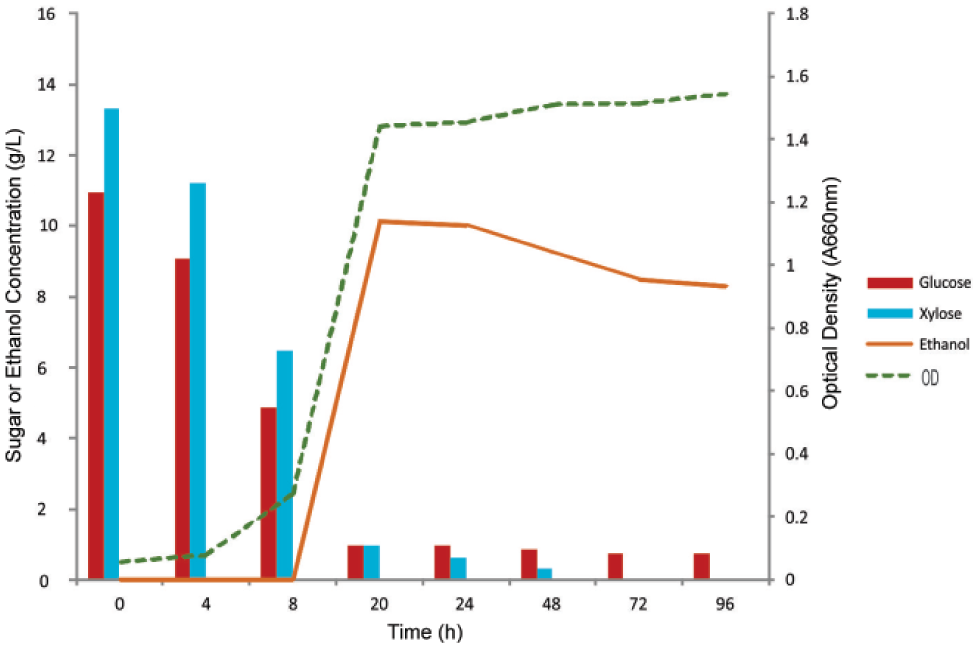
Growth, substrate consumption, and ethanol production of INVSc1 YAC4-SUMO-XI-XKS during fermentation experiments using hydrolysate of spent coffee grounds as a carbon source.
Because it has been observed that when glucose-xylose mixtures are incubated under either aerobic or anaerobic chemostat culture conditions, glucose is completely consumed, xylose consumption is incomplete, and significant concentrations of xylulose accumulate. 39 To investigate whether reactions occurring after xylulokinase were controlling ethanol production from xylose, Kuyper et al. 39 constructed a strain that overexpressed all enzymes required for conversion of xylose into fructose-6-phosphate and glyceraldehyde-3-phosphate. The overexpressed enzymes were xylose isomerase, xylulokinase, ribulose 5-phosphate isomerase, ribulose 5-phosphate epimerase, transketolase, and transaldolase. In addition, the nonspecific aldose reductase encoded by GRE3, which mediates production of xylitol, was deleted. The resulting strain grew anaerobically on 2% xylose in synthetic medium without any nondefined mutagenesis or selection. Xylulose formation was absent and xylitol production was negligible. During anaerobic batch cultivation, the specific growth rate of this strain on 2% xylose was 0.09/h, and ethanol yield was 0.43 g ethanol/g xylose calculated based on the ethanol concentrations deduced from CO2 production. In anaerobic batch cultures of mixtures of 2% glucose plus 2% xylose, both sugars were completely consumed, but glucose was the preferred substrate. Xylose consumption was initiated after approximately 80% of the glucose had been consumed, and during this phase, growth was not exponential and the specific rate of xylose consumption decreased over time. Alcohol yields on the two sugars were comparable. The specific growth rate on 2% xylose + 2% glucose during anaerobic batch cultivation was 0.25/h deduced from the glucose consumption phase, and ethanol yield was 0.43 g ethanol/g (glucose + xylose) based on the ethanol concentrations deduced from CO2 production. The authors noted that the true simultaneous utilization of glucose and xylose depends on a variety of parameters that are intrinsic to the regulatory circuits in the cell and that whether or not it will be achieved, it is evident that higher rates of xylose utilization by further improved strains will make alcoholic fermentation of sugars in plant biomass a realistic enterprise. 39
A study by Matsushika et al. 40 to determine whether the dynamic metabolism of a recombinant glucose/xylose-cofermenting S. cerevisiae strain genetically engineered with chromosome-integrated genes encoding Pichia stipitis xylose reductase and xylitol dehydrogenase and S. cerevisiae xylulose kinase was affected differently when using xylose as the substrate than when using glucose alone or mixed glucose plus xylose as substrates. Their results demonstrated that when xylose instead of glucose was metabolized by this strain, glycolytic metabolites, including 3- phosphoglycerate, 2- phosphoglycerate, phosphoenolpyruvate, and pyruvate, were dramatically reduced, while conversely, most pentose phosphate pathway metabolites such as sedoheptulose 7- phosphate and ribulose 5-phosphate were greatly increased, suggesting that the low metabolic activity of glycolysis and the pool of pentose phosphate pathway intermediates are potential limiting factors in xylose utilization. 40
These results of our work suggest that introduction of the genes for heterologous expression of XI and for overexpression of endogenous XKS in a synthetic cassette in a yeast artificial chromosome has improved xylose utilization by the INVSc1 yeast strain. It is clear that more work is still needed to understand the factors affecting the utilization of xylose by S. cerevisiae. Our study demonstrates that artificial chromosome-expressing genes to enhance xylose utilization can be stably introduced into an ethanologenic yeast strain without affecting the integrity of the host strain. The assembly and transformation processes are designed for use in an automated format, and the system provides a potential synthetic biology platform for one-step construction of a yeast artificial chromosome containing a cassette for expression of multiple genes to further improve industrial yeast strains. These genes could include genes for an entire metabolic pathway,39,41 novel genes identified using genome-wide transcription analysis, 42 genes from S. cerevisiae xylose-using strains, 4 genes selected from a library of strains each expressing a different native S. cerevisiae gene ORF, 10 or genes for high-value bioproducts. 37 The major advantages of the artificial chromosome system are that it can incorporate very large DNA inserts and can stably transform yeast strains with genes engineered to express proteins that confer improved or additional desired characteristics while maintaining the original fermentation capabilities of the host strain.
Footnotes
Acknowledgements
We acknowledge the assistance of Nathane Orwig in obtaining the PCR primers and of Eric Hoecker in conducting the high-performance liquid chromatography experiments.
Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the United States Department of Agriculture. The USDA is an equal opportunity provider and employer.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.