
Abstract
Drug-induced liver injury (DILI) is a leading cause of drug attrition. Significant and well-documented differences between animals and humans in liver pathways now necessitate the use of human-relevant in vitro liver models for testing new chemical entities during preclinical drug development. Consequently, several human liver models with various levels of in vivo–like complexity have been developed for assessment of drug metabolism, toxicity, and efficacy on liver diseases. Recent trends leverage engineering tools, such as those adapted from the semiconductor industry, to enable precise control over the microenvironment of liver cells and to allow for miniaturization into formats amenable for higher throughput drug screening. Integration of liver models into organs-on-a-chip devices, permitting crosstalk between tissue types, is actively being pursued to obtain a systems-level understanding of drug effects. Here, we review the major trends, challenges, and opportunities associated with development and implementation of engineered liver models created from primary cells, cell lines, and stem cell–derived hepatocyte-like cells. We also present key applications where such models are currently making an impact and highlight areas for improvement. In the future, engineered liver models will prove useful for selecting drugs that are efficacious, safer, and, in some cases, personalized for specific patient populations.
Introduction
Drug development is a very expensive venture, now costing ~$3 to $5 billion and 12 to 15 years to launch a single drug into the market.1,2 Lead candidate compounds typically undergo ADMET (absorption, distribution, metabolism, excretion, toxicity) characterization in vitro and in vivo (in animals) before entering human clinical trials ( Fig. 1 ). However, almost 90% of compounds that pass through preclinical drug screening end up failing during clinical trials, and one-third of these failures have been attributed to toxicity. 3 Furthermore, ~90% of withdrawals of drugs from the marketplace are due to toxicity issues. Of such toxicities, drug-induced liver injury (DILI) is the most common cause of acute liver failures in the United States alone and is a leading cause of both the prelaunch and postmarket attrition of pharmaceuticals. 4 For instance, DILI accounts for ~40% of the drugs that fail during clinical trials and has been linked to ~1000 marketed drugs. 5
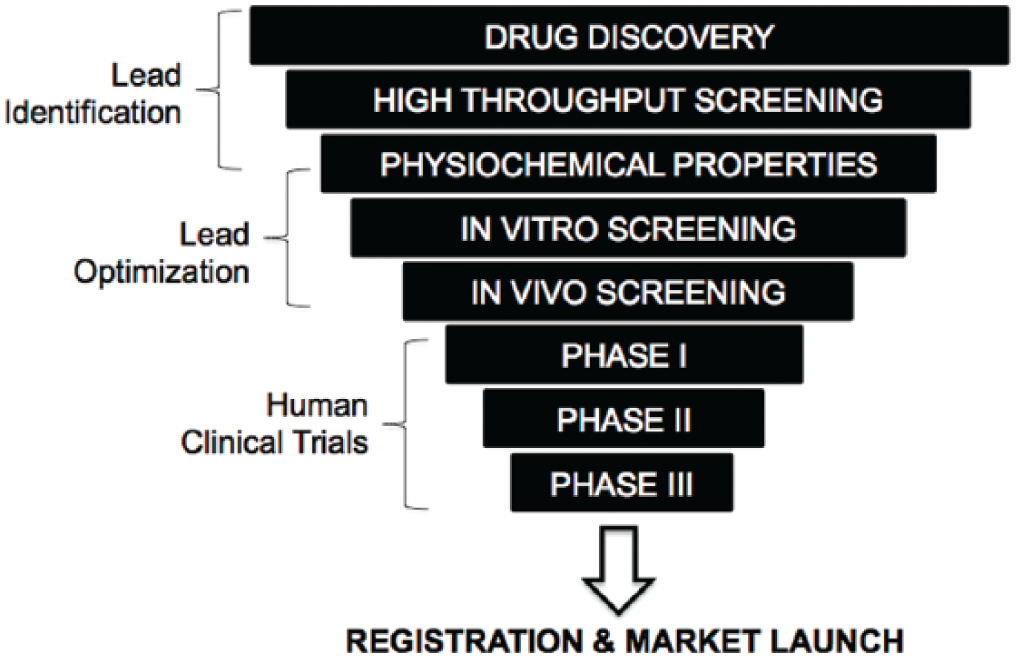
The process of drug development. The funnel schematic represents the fact that millions of chemicals created via combinatorial chemistry enter the process, but typically only one drug gets launched into the marketplace ~12 to 15 years later, costing ~$3 to $5 billion.
While effective in many cases, testing drugs on animals is not a fail-safe paradigm, especially for prediction of human DILI. About 50% of drugs known to cause human liver injury were not identified as toxic to the liver based on nonclinical animal testing. 6 Such lack of concordance is likely due to the significant differences in liver pathways (i.e., drug metabolism enzymes) between animal and human livers.7–9 In addition, preclinical safety evaluation studies are conducted in young animals with limited genetic diversity under controlled nutritional and housing conditions. However, it is known that risk factors in human patients include disease, sex, age, comedications, nutritional status, innate immune system activation, physical activity, and genetic predisposition. 10 Thus, there are increasing pressures on regulatory agencies and the pharmaceutical industry to find more effective ways to understand and predict human response to drugs in preclinical settings prior to the initiation of clinical trials.
An important route to accomplish the aforementioned goal has been increased preclinical utilization of human-relevant in vitro liver models, such as microsomes, cancerous cell lines, primary human liver cells, and liver slices.11–13 While these models have already been used to reduce risks in drug development, there remains a need for more sophisticated in vitro model systems that better capture liver physiology with which to probe and identify pathways that are perturbed following acute and chronic exposure to drugs at clinically relevant concentrations. Furthermore, understanding how the physiologic interconnections between organ systems affect overall drug disposition, efficacy, and multiorgan toxicities has now become more relevant than ever.
Different groups in both academia and the biotech industry are creating liver model systems using a variety of cell sources (i.e., primary, cell lines, stem cell-derived) that are more predictive of clinical outcomes than existing solutions. Here, we will describe the advances that are being made in designing such predictive human liver models. The use of engineering tools, such as protein micropatterning and microfluidics, to exercise better control over the microenvironment of liver cells is emphasized. Current and emerging applications of human liver models during the drug development pipeline are discussed. Finally, common strategies and challenges in this growing field are reviewed along with proposed validation schemes for in vitro engineered liver models.
Cell Sourcing Considerations
The liver is often described as the chemical factory of the body with over 500 functions. Some of these functions include protein synthesis (i.e., albumin, clotting factors), cholesterol metabolism, bile production, glucose and fatty acid metabolism, and detoxification/metabolism of endogenous (i.e., bilirubin, ammonia) and exogenous (i.e., drugs, environmental toxins) substances. Xenobiotics undergo three phases of metabolism and transport in hepatocytes ( Fig. 2 ). Phase I is the first-pass metabolism of lipophilic compounds into water-soluble metabolites for the purpose of removal from the body, and such reactions are mainly catalyzed by the cytochrome P450 family (CYP450) of enzymes specializing in oxidation and reduction reactions. Phase II enzymes conjugate highly polar molecules such as glucose, glucuronic acid, sulfate, or glutathione to xenobiotics and/or their metabolites. Although phase I and II metabolism in the liver is typically referred to as “metabolic detoxification,” many xenobiotics are metabolized into pharmacologically active or toxic compounds. 14 In phase III of drug disposition, highly polar metabolites are transported out of hepatocytes via transporters into the bile through the bile canaliculi or are released back into the blood for excretion via the kidneys.
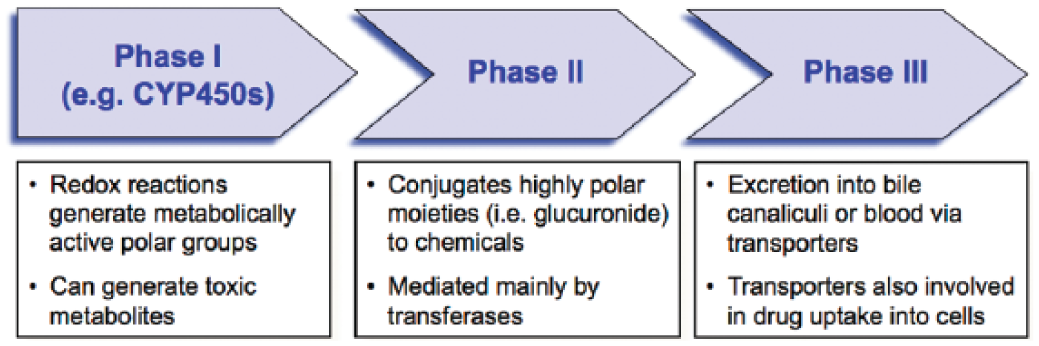
The three phases involved in metabolism and transport of drug/chemicals and their metabolites in the liver. Some of these enzymes are also found in other organ systems such as the intestine.
Microsomes, which are vesicle-like artifacts re-formed from pieces of the endoplasmic reticulum when cells are broken up, contain phase I enzymes and have proven very useful for targeted questions around profiling which CYP450s are relevant in metabolism of a given drug. 15 More recent research has focused on creating miniaturized arrays of spotted enzymes in gels to make the screening process higher throughput. 16 However, cell-free microsomes and other purified enzymatic systems lack the dynamic gene expression and cellular machinery required to be useful for drug toxicity and drug efficacy screening. In contrast to microsomes, precision-cut liver slices are the closest representation of an intact liver architecture with all the relevant cell types of the liver. While there have been some advances in culture of liver slices in microfluidic devices to prolong their lifetime, 17 ultimately this particular model suffers from a rapid (hours to days) decline in liver functions, which inhibits the possibility of chronic drug dosing. Liver slices also do not readily afford the opportunity to build engineered systems on demand from the “bottom up,” customized for specific applications and at the level of throughput required during drug development. Thus, we focus on cell-based culture models in this review. We refer the reader to other review articles on microsomes and liver slices. 12,18–20
While primary human hepatocytes (PHHs) represent nearly 80% of liver volume (60% of the total cell population) and perform most liver functions, they are surrounded by nonparenchymal cells (NPCs), which represent ~6.5% of liver volume (40% of the total cell population). The remaining liver volume consists of vascular and ductal networks. Major liver NPCs include liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs), Kupffer macrophages (KMs), biliary epithelial cells (cholangiocytes), and pit cells (intrahepatic lymphocytes or natural killer cells). These NPCs contribute to the support and regulation of hepatic growth, functions, and in some cases diseased phenotype via production of paracrine factors. 21 Thus, isolated liver cells, with their diverse functions, are often required to generate culture systems that can provide an integrated assessment of drug disposition, toxicity, and efficacy for liver diseases.
Animal Hepatocytes Relevant for Drug Screening
The pharmaceutical industry in conjunction with regulatory agencies such as the US Food and Drug Administration (FDA) are moving toward the use of human-relevant model systems for appraising drug effects. Major funding initiatives from the National Institutes of Health (NIH) and the Defense Advanced Research Projects Agency (DARPA) to develop integrated microphysiological systems using human cells are further aiding this movement. However, because animals are currently used for drug development per mandate from the FDA, the selection of the “appropriate” animal model to test the in vivo efficacy and toxicity of a given class of drugs remains important. Furthermore, studies seeking to understand the genetic basis of drug toxicity can be performed, at least initially, using genetically diverse animals (i.e., mice). 22 Thus, with the goal of reducing and refining studies in animals and to compare drug responses with human liver cells, building in vitro liver models using hepatocytes from different animal species is currently a worthwhile endeavor.7,23,24 Indeed, animal hepatocytes (i.e., mouse, rat, dog, monkey) are distributed alongside PHHs either in suspension or in plated formats by most commercial vendors (i.e., Life Technologies [Carlsbad, CA], BioreclamationIVT [Baltimore, MD], Triangle Research Labs [Research Triangle Park, NC], Corning Biosciences [Tewksbury, MA]).
The FDA requires both a rodent species and a nonrodent species for drug testing. Rats (i.e., Sprague-Dawley) and dogs (i.e., beagle) are the most popular animal species used during drug development due to their robust availability from a variety of commercial sources (i.e., Charles River Laboratories [Wilmington, MA], Covance [Princeton, NJ], Jackson ImmunoResearch Laboratories [West Grove, PA]). Monkeys are also used but can quickly become cost prohibitive, especially if used in the early stages of drug development. Moreover, comparisons of nine CYP450 enzymes in mouse, rat, rabbit, dog, micropig, and monkey liver microsomes to those derived from human livers revealed that, while no single species had enzyme activities close to human for all enzymes tested, different species could potentially be suitable for in vivo testing once it is determined which human CYP450 enzymes are involved in the metabolism of a drug. 25 Surprisingly, monkey was not always the most suitable species relative to human CYP450s, and in some cases, mouse was better (i.e., CYP1A). In another instance of defining relevance between humans and animals for behavior of specific drug classes, the woodchuck has been shown to be a better model for chronic human-like hepatitis infection and disease progression than other species and thus was used to study the antiviral activity and toxicity of fialuridine and analogue compounds. 26 Hepatocytes from pigs have similar biotransformation capacities as human hepatocytes for generating metabolites of select compounds, 27 which has implications not only for use of porcine hepatocytes in drug testing but as a source for cell-based therapies in the clinic (i.e., bioartificial liver devices). Fish hepatocytes have proven particularly useful for assessing the effects of human-excreted pharmaceuticals in water (i.e., effluent discharges from wastewater treatment plants) on fish populations. However, fish hepatocyte models are inadequate for prediction of drug effects on humans due to species-specific differences in drug metabolism pathways. 28 Accordingly, in vitro comparisons of drug metabolism across different animal and human liver models are necessary prior to selecting a species for FDA-required in vivo animal studies.
Some interesting developments in the use of nonhuman species for drug testing have been zebrafish whole embryos for testing the potential toxicity of new pharmaceuticals. 29 This model allows for the use of a whole organism with multiple organs present to evaluate the effects of drugs in a high-throughput format. While the zebrafish may very well bridge the gap between in vitro liver models and lower throughput testing in larger animals, further validation with a larger set of well-annotated drugs is needed to determine how liver pathways in zebrafish diverge from human livers. “Humanized” rodent models represent another exciting development. Rodent livers are either damaged and repopulated with replication-competent PHHs30,31 or PHHs are housed in a 3D synthetic scaffold and implanted in an ectopic site (i.e., intraperitoneal), leaving the rodent liver fully intact. 32 Human-relevant drug metabolites generated via the PHHs implanted in rodent livers could generate toxicity in another organ, thereby allowing assessment of safety of such metabolites. However, the presence of residual rodent hepatocytes coupled with the interaction of implanted PHHs and other rodent organs can present challenges in interpreting drug pharmacokinetics and organ toxicity data sets. Furthermore, live animal studies are inherently lower in throughput and variable from one animal to the next. In vitro models provide complementary tools in the early stages of drug development, where new drug manufacturing scale-up is limiting and higher throughput is necessary.
Human Hepatic Cell Lines
Cancer-derived or immortalized hepatocytes can be propagated in vitro over many passages, effectively constituting cell lines. Several hepatic cell lines have been used for drug development, including HepG2, HepaRG, Fa2N-4, and others.8,33,34 While providing for a reproducible and nearly infinite source of liver cells for building initial iterations of engineered devices as well as for drug testing, cell lines are widely accepted to lack the high levels of differentiated liver functions observed in PHHs.11,35 For instance, the SV40 large T antigen-immortalized Fa2N-4 cell line was found to have significantly lower expression of drug metabolism enzymes and uptake transporters than PHHs. Furthermore, Fa2N-4 cells were lacking an important nuclear receptor (constitutive androstane receptor or CAR), which limits the utility of these cells for drug-mediated enzyme induction studies to predict drug-drug interaction potential. 34
The reported sensitivities and specificities of cell lines for accurate detection of liver toxic drugs are conflicting. For instance, Atienzar et al. 36 reported a sensitivity of 80% for HepG2 cells using 40 known liver toxic drugs but a low specificity of 40% (i.e., high number of false positives) using 11 non–liver toxins. Gerets et al., 8 on the other hand, reported a sensitivity of only 6.3% for HepG2 cells using 16 liver toxic drugs with 100% specificity using five non–liver toxins. While HepG2 cells may provide an acceptable model for testing toxicity of parent drugs, they typically do not suffice for those drugs that are metabolically activated into toxic metabolites given the very low metabolic capacity of this cell line. This conclusion is consistent with another study that evaluated HepG2 cells alongside Huh7, SK-Hep-1, Hep3B, and HepaRG cell lines and showed a complete absence or much lower abundance of certain drug metabolism enzymes and transporters in hepatic cell lines compared with multiple PHH donors. 37 Transfecting HepG2 cells with an adenovirus expressing the major CYP450 enzymes involved in drug metabolism resulted in lower IC50 values of bioactivated compounds compared with dosing in untransfected controls. 38 However, drug metabolism and toxicity are often very complex, involving multiple enzymatic processes; therefore, the utility of this approach needs to be tested with transfection of additional proteins, and then validation of the resultant cells should be carried out by testing toxicities of a wider set of prototypical compounds.
The HepaRG cell line, which spontaneously differentiates in vitro into hepatocyte-like and cholangiocyte-like cells (representing the biliary epithelium of the liver), has emerged as a further differentiated and functional cell line relative to HepG2 and other cell lines. 39 In particular, their use in enzyme induction studies has been well documented. For instance, Gerets et al. 8 compared CYP450 induction at the gene expression and functional levels in both PHHs and HepaRG using three prototypical inducers and reached the conclusion that HepaRG could be used in CYP450 induction screening as a substitute or complementary to PHHs. Le Vee et al.40,41 showed that HepaRG have key sinusoidal and canalicular membrane transporters at both the transcript and functional levels, whereas HepG2 cells showed notable expression of fewer transporters. Szabo et al. 42 further showed that HepaRG could serve as a good model for evaluating the effects of drugs on the uptake of probe substrates and the potential for downstream cholestasis. Another group showed that HepaRG could be used as a substitute for PHHs for evaluating drug clearance. 43 Although CYP450 activities between PHHs and HepaRG varied, clearance rates of select drugs were similar between the two models. However, the sensitivity for drug toxicity detection in HepaRG was shown to be significantly lower (16% vs. 44%) than PHH cultures, 8 suggesting that HepaRG may not suffice for this particular application. Furthermore, as with all cell lines, ultimately HepaRG cells provide information on drug behavior in a single liver donor, which necessitates the complementary use of PHHs to obtain multidonor information.
Pluripotent Stem Cells
Embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) are capable of extensive self-renewal and can be differentiated into each of the three germ layers (endoderm, mesoderm, and ectoderm). Given their ability to be derived from adult somatic cells, iPSCs have revolutionized the availability of pluripotent stem cells for multiple applications ( Fig. 3 ). iPSCs were shown to be generated from human skin through the ectopic expression of select genes (i.e., Oct3/4, Sox2, c-Myc, Klf4, Nanog, Lin28).44–46 The use of nonintegrating episomal vectors allowed for the production of iPSCs free of viral vector and transgene sequences. 47 In addition, iPSCs have been generated via the addition of fewer genes in certain cell types, small molecules, recombinant proteins, adenoviruses or Sendai viruses, messenger RNA (RNA), recombinant proteins, and transient expression plasmids.48,49 Human iPSCs can mimic human ESCs in all aspects of pluripotency and could allow for the creation of donor panels that represent key polymorphic variants within a target population to help understand interindividual variability in drug responses (i.e., personalized medicine). Differentiated cells derived from stem cell sources could also provide a nearly unlimited supply of cells for (a) building sustainable and high-throughput drug screening platforms, (b) modeling organ development and diseases in vitro, and (c) enabling cell-based therapies such as cell transplantation, extracorporeal tissue devices, and implantable cell-laden engineered constructs.
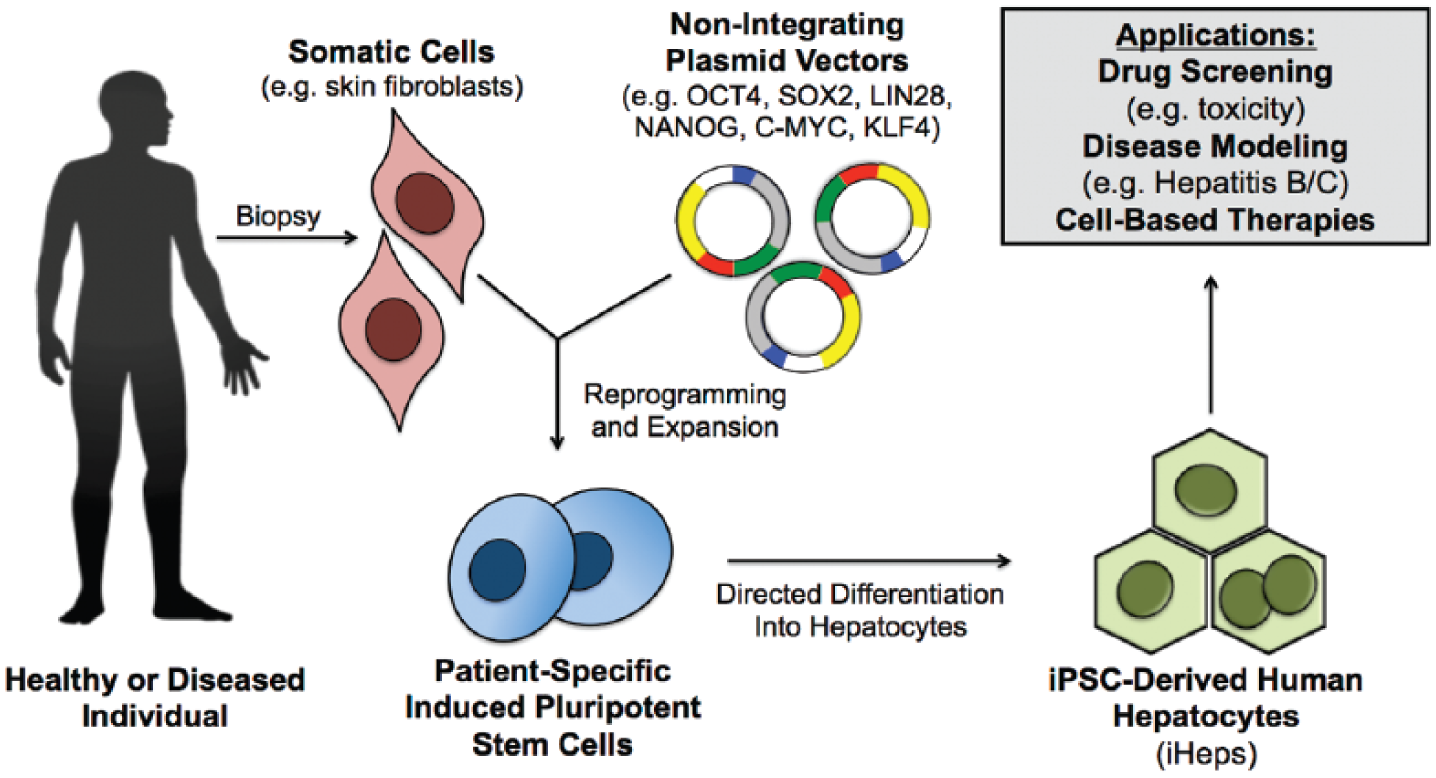
Protocols have been well established for the differentiation of either ESCs or iPSCs toward hepatocyte-like cells (iHeps).50–61 Typically, such protocols use cocktails of growth factors and small molecules to subject the cells to sequential differentiation steps such as endoderm induction, hepatic specification, hepatoblast expansion, and hepatic maturation. Recent protocols have eliminated the use of serum, feeder cell layers, the formation of embryoid bodies, and undefined reagents (i.e., tumor-derived Matrigel). Their removal not only maximizes the potential of these cells for future therapy but also provides more robust control over scaled-up manufacturing for drug screening applications. Some more recent approaches include stimulation with small molecules that can reduce fetal markers in iHeps, 62 deriving iHeps from PHHs to maintain the native epigenetic modifications, 52 and direct lineage reprogramming of fibroblasts into iHeps using key hepatic fate and maturation transcription factors.58,63
Despite advantages in sourcing and expansion of iHeps, some key issues prevent their routine and widespread utility in drug screening. In particular, functions in iHeps are often compared with PHH monolayers (>24 h in culture), which have severely degraded functions relative to levels found in vivo. Such suboptimal comparisons often bias the functional maturity in favor of iHeps. Regardless, it is widely accepted that in vitro differentiation protocols need to be improved to induce functions in iHeps closer to adult PHHs.51,64 Furthermore, an abundant supply of iHeps from different donors cultured in a reproducible culture format is not routinely available to investigators who may not be experts in iPSC biology but instead want to use the cells for drug screening. Alleviating these issues using engineering tools (i.e., microfabrication) is likely to help spur widespread use of iHeps in drug screening. The recent commercial availability of iHeps (i.e., Cellular Dynamics International, Cellectis) is greatly aiding in this effort.
Primary Human Liver Cells
PHHs, isolated from the human liver via collagenase perfusion, are widely considered to be the “gold standard” for use in constructing liver models for drug testing and other life science applications.11,12 A recent study showed that addition of an antioxidant, N-acetylcysteine, and replacement of collagenase with Liberase could increase the cell viability from both normal and diseased liver tissues. 65 PHHs have also been isolated from fibrotic and fatty livers, providing the ability to compare cells with a history of disease to control cells that do not display that particular disease phenotype. 66 Moreover, PHHs from liver biopsies extracted from living patients can provide a more viable source of PHHs than tissues from deceased and beating-heart donors. 67
Advances in cryopreservation methodologies have greatly facilitated the commercial availability of characterized PHH donor lots from several different vendors (i.e., Life Technologies, BioreclamationIVT, Triangle Research Labs). The use of cryopreserved PHHs affords several advantages that include convenient on-demand experimentation as opposed to the unpredictability in procurement of fresh cells; longitudinal studies in one donor, as opposed to significant interexperimental variability observed with the use of fresh PHHs from different donors; and comparisons across multiple donors for appraising the effects of donor characteristics on specific outcomes. 68 Characterization using gene expression profiling showed that isolation and cryopreservation did not significantly impair PHH gene expression relative to native liver tissue. 69 Functional characterization of fresh and cryopreserved PHHs from the same donors using prototypical drugs has also revealed significant similarity in responses. For instance, cryopreserved PHHs stored in liquid nitrogen for more than 1 year showed no significant decrease in viability or activity upon thawing, when compared with the initial thaw, which was carried out within a week of cryopreservation. 70 Furthermore, the activity of thawed cryopreserved PHHs was on average 94% of that of fresh PHHs for a range of drugs metabolized by major enzymes in the liver. However, in our experience, not all cryopreserved PHH lots attach to extracellular matrix (ECM)–coated tissue culture plastic with the same efficiency (and some do not attach at all) as their fresh counterparts. Thus, research in medium composition (i.e., energy precursors, antioxidants, oxygen levels) in which to incubate fresh PHHs for specific durations prior to cryopreservation remains an active area of investigation. Such preincubation has been shown to correlate with postthaw viability and attachment potential of PHHs to ECM-coated culture substrates.71–73
Under the appropriate culture conditions, both freshly isolated and cryopreserved PHHs that attach to ECM-coated surfaces can maintain high levels of phenotypic functions for several weeks in vitro, permitting investigations in chronic exposure to drugs and infectious diseases.12,74 PHHs have been used in a wide array of culture models, and their use for pharmaceutical drug screening has been expanding over the past two decades.11,12 While they are ultimately a limited resource, the high yields of PHHs from whole human livers (~5–10 billion), advances in cryopreservation, and extraction of PHHs from liver resections and biopsies in addition to whole livers have allowed robust use of PHH models in lead optimization stages of drug development. Thus, even as the above-mentioned cell lines and stem cell–derived hepatocyte-like cells provide complementary and cheaper tools for drug screening, we anticipate that PHHs will continue to play an important role in drug development for not only appraising the in vivo relevance of the hepatocyte-like cells and cell lines but also for providing an output closest to the native human liver.
NPCs of the liver, such as LSECs, KMs, HSCs, and cholangiocytes, also play roles in modulating hepatic functions in both physiology and disease states as well as hepatic responses to several drugs ( Fig. 4 ).21,75–77 Furthermore, some drugs cause cholestasis through disruption of the biliary tree in the liver instead of directly affecting the PHHs or liver NPCs. 78 While these nonhepatic cell types can be isolated from the rat liver with established protocols, commercial availability of human liver NPCs from multiple donors is still lagging behind PHH availability. KMs are available through at least two vendors (Life Technologies, BioreclamationIVT); however, given the low numbers of KMs in the liver (~1 KM for every 5-10 PHHs), availability of lots and number of vials per lot are not as robust as PHHs. In addition, the available lots need to be screened by the end user for attachment potential and functionality prior to banking for creation of long-term liver models. LSECs are difficult to propagate in vitro since they lose their prototypical fenestrae within a few days. 79 HSCs develop a myofibroblast phenotype rapidly in vitro, which is a hallmark of liver fibrosis.21,80 While modulating the compliance of a substrate has been shown to revert HSCs to a more quiescent state, 80 such substrates and techniques are not yet standard for drug screening applications. Even though NPC culture techniques are not as established as those of PHHs, as we illustrate in the next section, a few commercial vendors and academic groups have been co-culturing a variety of the aforementioned liver NPCs with PHHs in their respective model systems, and initial results so far have been promising.
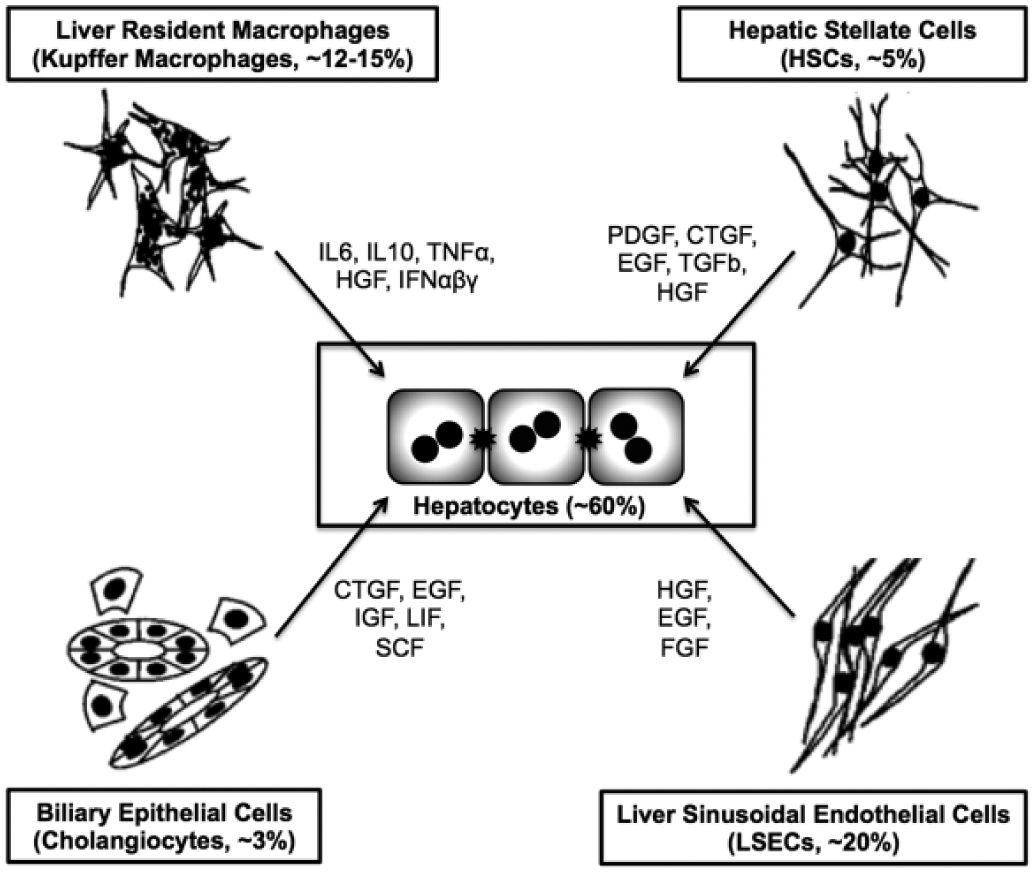
Effects of nonparenchymal cell types of the liver on parenchymal hepatocytes through paracrine secretions. Percentage values represent relative number of each cell type found in the human liver. Adapted from LeCluyse et al. 11
Engineered Systems for Culture of Liver Cells
The goal of any type of cell culture is to provide an in vitro microenvironment that can maintain phenotypic functions of cells as close to the native tissue as necessary for a specific downstream application ( Fig. 5 ). Such retention of an in vivo–like phenotype affords the investigator the ability to reliably and accurately investigate in vitro the detailed mechanisms underlying phenomena observed in living organisms. Issues of cost, throughput, and the ability to manipulate and assess cellular morphology and functions are also important, especially for drug screening applications. In the case of the liver, the phenotype of an isolated hepatocyte is highly sensitive to temporal and spatial presentation of microenvironmental cues. Thus, culture of hepatocytes in various formats has been carried out for several decades, and some very comprehensive reviews cover the historical perspective and advances that have been made in this field.11,12,15,81,82 Briefly, the most popular culture configurations are ones in which confluent monolayers of primary hepatocytes are attached to adsorbed or gelled rat tail collagen I (widely and cheaply available) and then overlaid with another gelled ECM, such as collagen or Matrigel, to create the so-called ECM “sandwich” culture model. This ECM sandwich slows down but does not prevent the “de-differentiation” (i.e., severe reduction in major liver functions, including those related to drug metabolism) of hepatocytes observed in simple confluent monolayers on adsorbed collagen.12,74,81 More recently, a few enhancements have been made to the sandwich technique, including seeding of endothelia on top of the overlay as well as use of chitosan/hyaluronic acid polyelectrolyte multilayers as an overlay instead of collagen to provide better control over the chemomechanical environment around hepatocytes.83–85 So far, effects of these enhancements have been limited to rat hepatocytes, and their translation to PHHs is pending. Nonetheless, these examples demonstrate that the sandwich model can be built upon to include additional cues that stabilize diverse functions of hepatocytes and liver NPCs. As we discuss in subsequent sections, the sandwich model is also being incorporated into bioreactors.
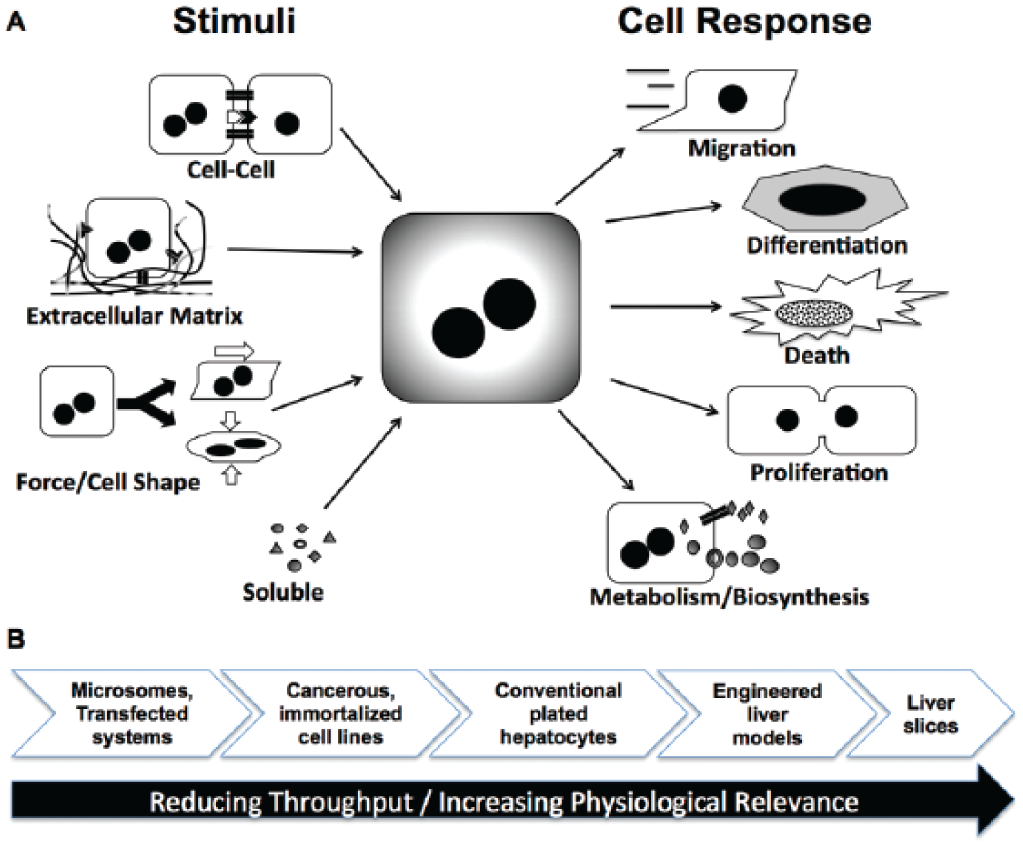
Effect of the microenvironment on cell fates. Some key microenvironmental cues, especially those that are relevant in the liver, are shown (
Other culture configurations include seeding hepatocytes directly on a gelled Matrigel layer to create small adherent spheroids. Spheroids can also be created via other means and have been shown to promote higher functions in hepatocytes compared with simple confluent monolayers on adsorbed collagen.86–88 Coculture with both liver- and non–liver-derived NPCs has been long known to induce functions in hepatocytes from different species. 82 More recently, a handful of groups have tested this so-called coculture effect on iHeps using non–liver NPCs such as murine embryonic Swiss-3T3 fibroblasts as well as a combination of MSCs (mesenchymal stem cells) and HUVECs (human umbilical vein endothelial cells).89,90 Liver-inspired complex ECM mixtures and ECM derived directly from the liver have also been shown to induce key hepatic functions.91,92 Finally, supplementation of culture medium with soluble factors, such as hormones (i.e., insulin, glucagon) and corticosteroids (i.e., dexamethasone or hydrocortisone), appears to be critical for any type of culture model; however, by themselves, culture medium supplements do not completely rescue the hepatic phenotype.11,15
The aforementioned microenvironmental cues have been superimposed onto hepatocyte cultures by many investigators in a “randomly distributed” manner. We refer the readers to other review articles that discuss a plethora of these conventional techniques.12,93 Here, we focus on those culture models that have been subjected to engineering tools, such as those adapted from the semiconductor industry and synthetic scaffolds for 3D tissue generation, to provide better control over the hepatic microenvironment, which has undoubtedly been shown to be important for optimizing liver functions in hepatocytes.74,94,95 Most of the models we discuss have also been miniaturized to various degrees for higher throughput drug screening. However, some engineered liver models are being developed to provide high-content information (as opposed to high-throughput screening) such that they can be used in later stages of drug development, prior to clinical trials. Finally, we emphasize PHH-based culture systems, designed to recapitulate in vivo human liver functionality as closely as possible, but also present examples of similar techniques for maturing iHep functions in specific cases.
2D Micropatterned Models
Conventional monolayer cultures are generated by randomly seeding hepatocytes onto substrates coated homogeneously with adsorbed collagen or other types of ECM. In contrast, through the use of selective surface modification, microfabrication tools allow generation of heterogeneous surfaces that offer control over cell-ECM and cell-cell interactions with micrometer precision.96,97 A variety of such micropatterning techniques are reviewed in other articles.98,99 Briefly, photolithography allows patterning of photoresist (light-sensitive polymer) onto a silicon or glass wafer. PDMS (polydimethylsiloxane), a biocompatible silicone rubber, can be cast onto the wafer to yield a stamp (termed soft lithography) for subsequent use in the microfluidic delivery of proteins and cells or in microcontact printing of organic molecules (i.e., proteins) onto substrates ( Fig. 6 ). The overall goal of using micropatterning and microfluidics is to modulate behavior of cells by precisely controlling their microenvironment.
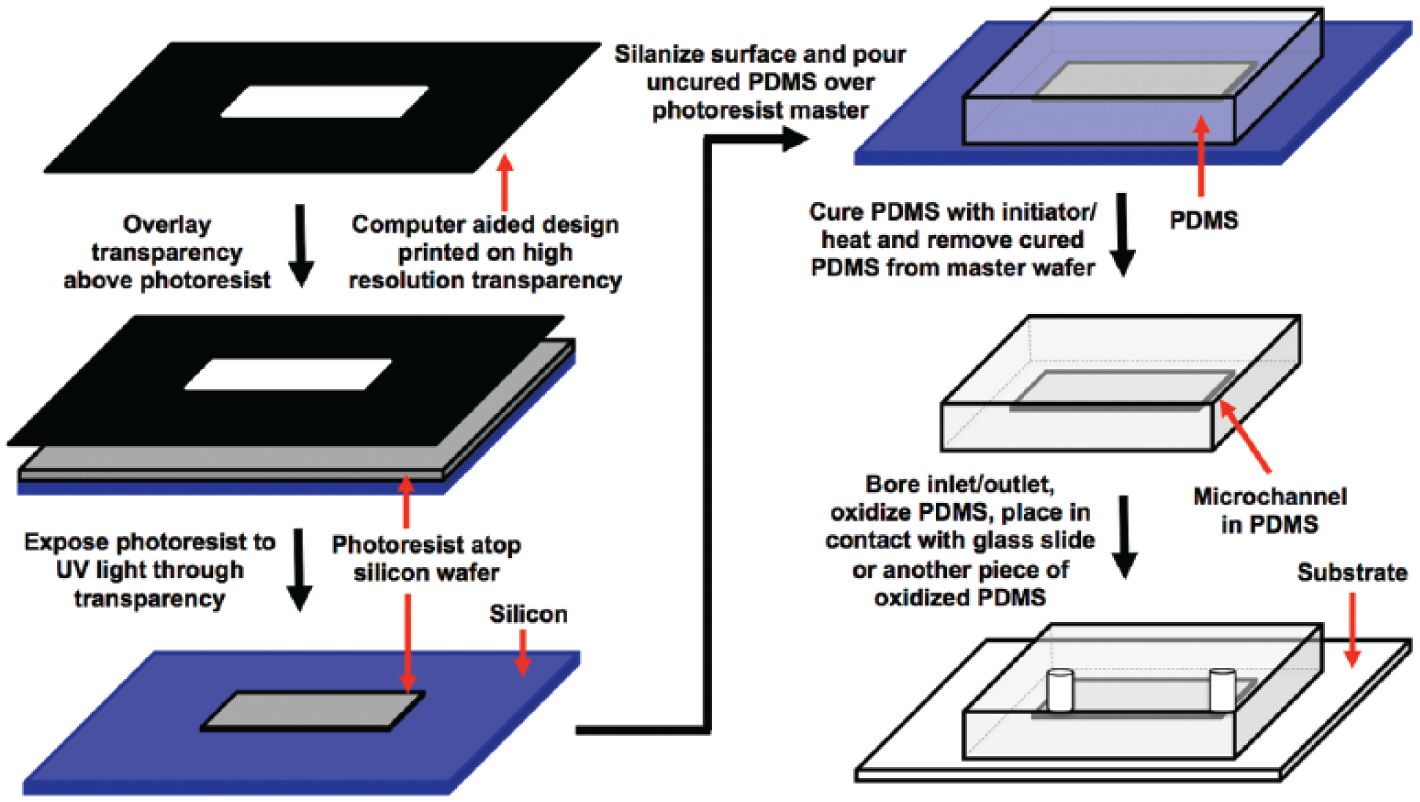
Creation of microfluidic devices. A photolithographic process is used to first create a pattern of a photoresist on a silicon wafer. Then, soft polymers such as polydimethylsiloxane (PDMS) are cast on the wafer to generate a stamp. This stamp can then be irreversibly bonded to a basement substrate (i.e., glass, PDMS, or acrylic) to create microfluidic channels for perfusion of cultures. 205
Microfabrication tools have been used extensively to investigate biological phenomena in different model systems.98,100 In the case of the liver, Singhvi et al. 101 demonstrated culture of rat hepatocytes on patterned self-assembled monolayers (SAMs) of alkanethiols with adsorbed fibronectin, which were surrounded by nonadhesive polyethylene glycol SAMs to keep cells from migrating off the fibronectin-coated domains. The authors demonstrated increased differentiated function of hepatocytes (albumin secretion) and reduced DNA synthesis as a marker of de-differentiation by constraining spreading of the hepatocytes on adhesive domains of different sizes. However, interactions between hepatocytes alone were not sufficient to rescue their phenotype in vitro over prolonged times in culture (weeks).
Bhatia et al. 96 then extended the aforementioned patterning to micropatterned coculture (MPCC) of rat hepatocytes with supportive NPCs. The development of these MPCCs was inspired by studies by Guguen-Guillouzo et al., 102 who demonstrated transient stabilization of some PHH functions upon co-cultivation with a liver-derived epithelial cell type. However, with the random seeding of the two cell types, it was not possible at that time to explore the role of controlled cell-cell interactions on hepatic functions. Thus, Bhatia et al. employed photolithography to first micropattern 2D islands of primary rat hepatocytes attached on adsorbed collagen and then surrounded those islands by 3T3-J2 murine embryonic fibroblasts, a cell type that has also been used in coculture with keratinocytes. 103 Photolithography allowed tuning of homotypic interactions between hepatocytes alone while keeping cell numbers constant across the various patterned configurations. These studies revealed that cell-cell contact played a critical role in modulating hepatocyte functions by several fold, likely due to cadherin interactions as observed in other studies. 81 As in studies by Guguen-Guillouzo et al, 102 physical contact with stromal cells (i.e., fibroblasts) was required in MPCCs to significantly augment both the magnitude and longevity of the hepatocyte phenotype by several weeks as opposed to a declining phenotype in pure micropatterned hepatocyte cultures. 82 Subsequently, Khetani et al. 104 conducted a functional screen, which revealed that 3T3-J2 cells induced optimal functions in hepatocytes from multiple species (rat, human) compared with other 3T3 clones (i.e., NIH-3T3, Swiss-3T3, L1-3T3).
The MPCC platform and associated protocols were subsequently modified by Khetani and Bhatia 74 for freshly isolated PHHs as these cells became routinely available via several commercial sources at the turn of this century. However, a different balance of homotypic and heterotypic interactions with 3T3-J2 fibroblasts was needed to induce optimal functions in PHHs relative to rat hepatocytes. Next, soft lithographic techniques based on PDMS were developed to allow repeated creation of MPCCs in miniaturized multiwell formats for higher throughput screening. In particular, PDMS stencils were initially used to pattern ECM, and then a PDMS gasket was used to provide 24 distinct wells for drug dosing over the MPCCs. 74 However, since PDMS is a porous and hydrophobic material, drugs and other hydrophobic molecules in culture medium (i.e., proteins) had a tendency to get soaked up in the polymer, thereby reducing the effective molecule concentration available to the cells. Thus, a new process was developed in which a PDMS stamp is used as a mask to protect certain regions of an ECM coat from being ablated by oxygen plasma. Once the ablation is complete, the cells are seeded on the remaining ECM patterns in standard tissue culture polystyrene industry-standard 24- and 96-well plates that do not contain PDMS ( Fig. 7 ).74,105 Furthermore, MPCC culture protocols were adapted by investigators at Hepregen Corporation (Medford, MA) to use different batches of cryopreserved PHHs for on-demand device creation for pharmaceutical drug testing and other life science applications.106,107
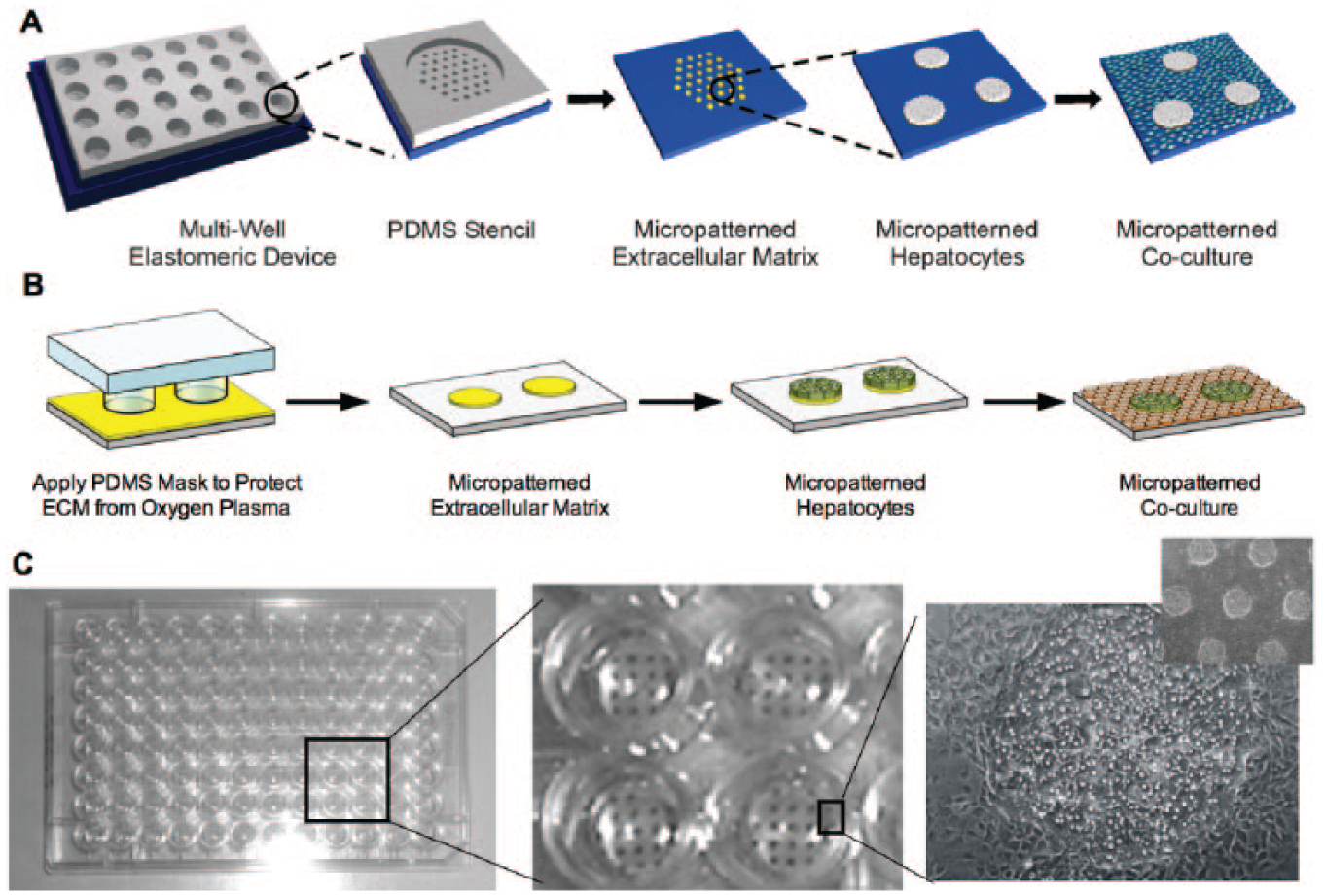
Soft lithographic processes to create micropatterned cocultures. Polydimethylsiloxane (PDMS) stencils created using lithography can be used to first pattern extracellular matrix or ECM (i.e., collagen) onto tissue culture polystyrene. Hepatocytes attach only to the ECM domains. Cells that have not attached to the ECM domains are washed away a few hours later, and on the next day, nonparenchymal cells are seeded and attach to areas not occupied by the hepatocytes (
Today, functionally optimized MPCCs contain hepatocytes organized in ~500-µm circular islands (~200–250 cells/island), spaced 900 to 1200 µm apart center-to-center depending on the application, and then surrounded by 3T3-J2 fibroblasts. Such circular architecture has been found to remain intact with respect to the fidelity of patterning, hepatic morphology, gene expression, and liver functions for 4 to 6 weeks for PHHs 74 and up to 10 weeks for rat hepatocytes. 24 MPCCs have been extensively validated for use in predictive drug testing. For instance, MPCCs created using PHHs have been shown to be ~75% predictive of clinical outcomes for drug metabolite and DILI profiling as opposed to <50% sensitivity in standard culture systems.106,107 Furthermore, MPCCs have been used for the study of hepatotropic pathogens that will be discussed in the infectious disease section of this article.108–110 MPCCs allow simultaneous evaluation of drug metabolism, toxicity, and efficacy of candidate compounds in the same hepatocytes due to maintenance of high levels of drug metabolism enzymes (i.e., CYP450s) and drug transporters localized on the proper membranes (basolateral, apical). 108
Other groups have also created micropatterned cocultures of hepatocytes and stromal cell types. For instance, Zinchenko et al. 111 created micropatterned cocultures of rat hepatocytes with Kupffer macrophages using both photolithographic and soft lithographic (stencils) techniques and found better liver functions for 10 days in culture. However, given the decline in hepatic functions over 10 days, it appears that 3T3-J2 fibroblasts induce higher and more stable functions in rat hepatocytes than Kupffer macrophages alone. A recent modification to MPCCs, albeit only with rat hepatocytes so far, has been to use PDMS stencils to culture hepatocytes on top of micropatterned fibroblast colonies (i.e., layered) and compare functions with the coplanar configuration. 112 The authors found that increasing the hepatocyte-fibroblast interactions via layering improved several liver functions and allowed more uniform albumin staining in the entire hepatocyte island than in the coplanar configuration. In another technique, Nahmias et al. 113 developed a laser-guided direct writing system to pattern hepatocytes and endothelial cells “on the fly” with micrometer precision on arbitrary matrices, including soft gels. While this technique affords the greatest flexibility in patterning cell types compared with photolithographic or soft lithographic techniques described above, it is a serial process that can take ~7 h to create a handful of devices.
More recently, we have shown that the MPCC platform, when combined with a Matrigel overlay (i.e., hybrid of MPCC and ECM sandwich techniques), is useful to further mature iHeps toward a more adult-like PHH phenotype and maintain functions for several weeks in culture for chronic drug dosing studies. 114 In particular, commercially available iHeps (iCell Hepatocytes by Cellular Dynamics International) maintained elevated levels of albumin and urea production, CYP450 gene expression and enzyme activity, and sensitivity to prototypical drugs when cultured in our so-called iMPCC platform compared with pure iHep cultures. 114 Moreover, global gene profiling revealed remarkable similarities in the hepatic gene expression and establishment of liver-specific gene regulatory networks in iMPCCs compared with freshly isolated PHHs and those stabilized in MPCCs.
One of the challenges in using any type of multicellular culture is the ability to separate signals from specific cell populations. For instance, any time a non–liver-specific biomarker is assessed in MPCCs, fibroblast-only controls typically need to be carried out to ascertain hepatocyte-specific responses. That being said, since MPCCs are a 2D monolayer of cells, they are compatible with high content imaging, which is useful to determine the effects of drugs and other perturbations on hepatocytes and nonparenchymal cells in the same well.62,115 However, the use of imaging is restricted to the availability of fluorescent probes for functions of specific organelles. A more robust strategy to assess responses of different cell types involved the development of a mechanically actuated substrate to physically separate the two cell types following contact for downstream assessment of phenotype or gene expression of each cell type separately. 116 Thermo-responsive substrates have also been used to create micropatterned cocultures with better control over placement and subsequent removal of specific cell types.117,118 However, such “dynamic” cell culture substrates lead to a reduction in throughput for drug testing, and specific training of personnel on handling of the specialized devices is needed. Nonetheless, they are very useful to evaluate the mechanisms underlying heterotypic cell-cell interactions as well as for applications in regenerative medicine.
Even though we know that non–liver NPCs, such as murine embryonic 3T3-J2 fibroblasts, express and secrete molecules present in the liver (i.e., T-cadherin, vascular endothelial growth factor [VEGF], ceruloplasmin),104,119 it would be ideal to replace the non–liver NPCs altogether with molecules that stabilize the hepatic phenotype. Such replacement would allow assessment of hepatic responses without devising strategies to separate the fibroblast-specific signals. Using the aforementioned mechanically actuated substrates, Hui et al. showed that contact with fibroblasts was necessary for ~18 h, followed by stimulation of hepatocytes with secreted fibroblast factors that are likely to be labile such that the fibroblasts need to be present close to the hepatocytes (<400 microns) to enable a complete rescue of the hepatic phenotype. 116 Thus, fibroblast conditioned medium transfers from adjacent wells are unable to stabilize the hepatic phenotype. The small proteoglycan, decorin, and truncated cadherin (T-cadherin) were shown to be produced by the 3T3-J2 fibroblasts and induced functions in primary hepatocytes when presented as purified proteins mixed in with collagen.104,119 However, neither of these molecules completely rescued the hepatic phenotype, suggesting that other molecules are important in stabilizing hepatocytes. Studies are now ongoing to further elucidate the molecular mechanisms under the “coculture” effect.
Despite constituting a significant advance in the long-term culture of PHHs, current versions of MPCCs do not include other liver NPCs (i.e., LSECs, KMs, HSCs) that are known to modulate PHH functions in both physiological and disease states (i.e., drug toxicity).21,75–77 Furthermore, PHHs are cultured on rat tail collagen I instead of liver-inspired complex mixtures of human ECM proteins, which can be difficult to source in large quantities and are considerably more expensive than rat tail collagen I. Since MPCCs are built “bottom up” from individual components, they can be used as a base platform on which to engineer additional liver-specific microenvironmental cues that improve sensitivity for prediction of clinical drug outcomes. Thus, we and others are now working to create the next-generation MPCC model that builds in such complexities for more targeted questions. Nonetheless, the successful application of MPCCs for drug screening and infectious disease applications and its robust commercialization by Hepregen Corporation as HepatoPac for pharmaceutical drug screening constitutes an important step in the use of more sophisticated human liver models for obtaining better prediction of clinical drug outcomes than previously possible.
Even with sourcing limitations, published data indicate that it is possible to keep some of the liver NPCs functional in vitro for a few days to weeks.79,84,120 Interestingly, the various liver cell types affect each other’s stability in vitro. For instance, a tri-culture of hepatocytes, LSECs, and 3T3-J2 murine embryonic fibroblasts on the aforementioned mechanically actuated dynamic substrates was better able to maintain the phenotype of hepatocytes and fenestrate of endothelial cells as opposed to coculture between hepatocytes and endothelia alone. 79 Such a result suggests that LSECs benefit from being in coculture with 3T3 fibroblasts or stable hepatocytes (vs. declining ones) or both. Nahmias et al. 113 showed that hepatocytes migrated toward and adhered to endothelial vascular structures formed on Matrigel. However, this sinusoid-like structure collapsed after 10 days in culture unless some fibroblast-like cells were also present, in which case the tri-culture functioned for several months. Another group has demonstrated that coculture with freshly isolated LSECs was able to decrease HSC activation, but coculture with LSECs that had lost their differentiated state had no effect on HSCs. 121 While the proof of concept for complex cocultures has been demonstrated, reproducibility across devices with respect to differential growth, evolving gene expression, and in vitro adapted functions of the different cell types needs to be further appraised prior to use of these cells in large-scale device manufacturing.
3D Static Spheroid Models
Culture of hepatocytes into self-assembled 3D spheroids/aggregates has been carried out extensively on nonadhesive surfaces or ones that are highly compliant such that hepatocytes cannot fully spread out.12,87,122,123 The simplest examples of such models are to culture spontaneously forming hepatocyte aggregates on gelled Matrigel or nonadhesive bacteriological plates. Overall, culture of hepatocytes into spheroids has been shown to improve several categories of hepatocyte functions, likely due to establishment of homotypic cell-cell contacts and presence of key ECM components within and around the aggregates. Culture of iHeps in spheroids has also been shown to improve their liver maturation. 124 Here, we will focus on spheroid technologies that have been developed for drug screening instead of bioartificial clinical devices.
One key challenge with spontaneous formation of spheroids on nonadhesive plates has been the inconsistent size distribution that results in necrosis within the centers of large (>200 micron) spheroids due to diffusion limitations of key nutrients and oxygen. To mitigate such challenges, scaffolds and channels have been used to direct the assembly of the spheroids and to shorten the time required for spheroid formation by facilitating intercellular contact. For instance, spheroids of rat hepatocytes can be created by seeding cells into 24-well plates (microspace cell culture plate; Kuraray Co, Tokyo, Japan) having a specific spheroid-forming microarchitecture at the bottom of each well (grids of specific depth). 125 Another type of 96-well plate, available as Cell-able, (Cosmo Bio USA, Inc., Carlsbad, CA) allows semisphere-shaped and uniform-sized hepatocyte spheroids to form on ECM-coated adhesive domains surrounded by a nonadhesive coating, which are patterned using photolithography. 87 Liu et al. 126 created micropatterned tri-cultures of hepatocytes, fibroblasts, and endothelial cells on electrospun fibrous mats of PELA (poly(ethylene glycol)-poly(DL-lactide)) and Lac-PLA (lactosylated poly(DL-lactide)). These mats aided in the formation of hepatic spheroids and capillary-like endothelial structures. This tri-culture model enhanced liver functions, particularly albumin secretion, urea synthesis, and CYP450 activities.
A few spheroid-based liver culture models have made it into the commercial landscape. For example, in the RegeneMed (San Diego, CA) platform, liver NPCs (stellate cells, Kupffer macrophages, endothelia) are seeded onto a 3D porous nylon scaffold followed by seeding of hepatocytes (rat or human). 127 Human hepatic functions—in particular, secretion of albumin, fibrinogen, transferrin, and urea—were maintained for up to 3 months. In addition, these 3D liver cocultures maintained drug-mediated induction of CYP450s, formed bile canaliculi-like structures, and responded to inflammatory stimuli. This model detected clinically relevant drug toxicity, including species-specific drug effects, with higher sensitivity than pure hepatocyte monolayers. However, with such complex cultures containing multiple cell types, it is not always trivial to control the evolution of the model over time. Whole-genome microarray analysis revealed that after ~4 weeks, the model did not resemble an in vivo liver at least at the gene expression level, even though some hepatic functional markers were found to be stable past 4 weeks. 120 This type of result underscores the critical need to extensively characterize and validate engineered liver models over time using a variety of markers, including global gene expression profiling. Such analyses are necessary to carry out in several batches of the engineered tissues to demonstrate reproducibility and high quality prior to routine implementation in pharmaceutical practice. Nonetheless, the RegeneMed model was one of the first commercially available models to demonstrate the proof of concept that highly complex multicellular cultures can be created and that their development, characterization, and validation for drug testing should be pursued further.
The hanging-drop strategy by InSphero (Schlieren, Switzerland) allows hepatocytes (primary rat and human, HepG2) and NPCs (Kupffer macrophages, endothelia) to form controlled size microtissues in a specialized plate, which are subsequently transferred to another multiwell plate for drug testing ( Fig. 8 ). 86 Hepatocytes in these microtissues have been shown to be viable and secrete albumin for 30 to 35 days in vitro. Published drug validation data showed dose-dependent toxicity of acetaminophen, diclofenac, and trovafloxacin. Trovafloxacin toxicity, in particular, was sensitive to lipopolysaccharide (LPS)–mediated stimulation of Kupffer macrophages, a finding consistent with other studies in 2D monolayers. 128 While the consistency in size distribution of the microtissues is impressive (i.e., 253 ± 7.4 microns), further drug validation data will be needed on this model system to better appraise its advantages for drug screening over 2D cocultures. Regardless, one great advantage of the hanging-drop technique is that it is applicable to different types of cells, which helps to standardize some of the upstream manufacturing protocols to create different tissues. Indeed, Frey et al. 129 recently presented a platform featuring high flexibility in arrangements and interconnections between microtissues for different tissue types, which are formed in parallel on the same microfluidic chip. Control over liquid flow through the hanging drops enabled supply of nutrients, compounds, and intertissue metabolic communication. Such a platform is an encouraging step forward in the development of organs-on-a-chip that could be used to obtain a systems-level (i.e., multitissue) assessment of drug effects.
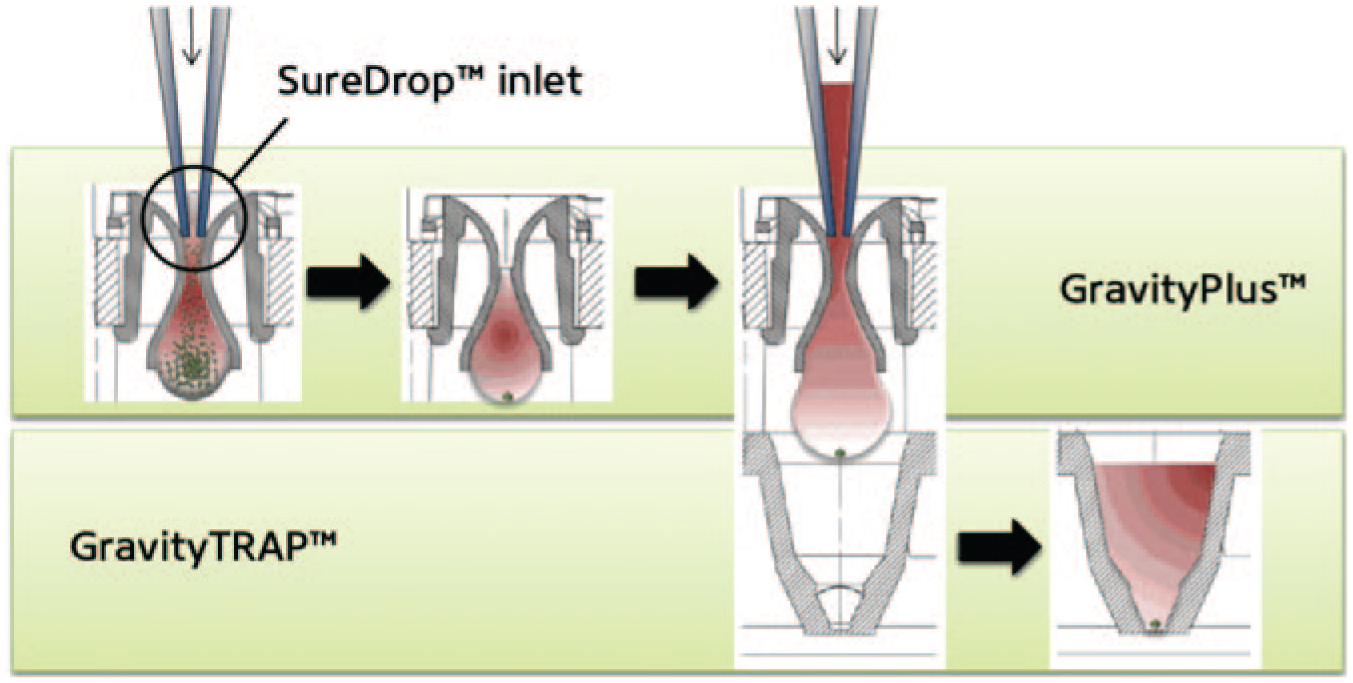
The InSphero strategy to create multicellular spheroids. The GravityPLUS 96-well plate allows a single microtissue to form in each drop. Once the microtissues are formed, they are transferred to a GravityTRAP plate. A proprietary nonadhesive coating allows for long-term culturing of microtissues without attachment. 86
Another promising strategy to create liver cell organoids is based on 3D bioprinting commercialized by Organovo (San Diego, CA). 130 One advantage of this technique is that printing in different locations can position different cell types relative to each other. For instance, liver NPCs (endothelia, stellate cells) were positioned in defined locations relative to hepatocytes, creating a compartmentalized architecture. Microvascular networks were observed within the 3D tissue as well as formation of tight intercellular junctions between hepatocytes. These 3D liver tissues were shown to display liver functions such as albumin production (five to nine times greater on a per-cell basis than matched 2D controls) and drug-mediated induction of CYP1A2 and CYP3A4 activities (unpublished data). More recently, these 3D liver organoids were shown to detect the toxicity of a drug that had previously been deemed safe in preclinical animal studies but ultimately caused liver damage in human patients. However, these customized 3D liver tissues need further characterization and validation with larger drug tests prior to full-scale adoption by the pharmaceutical industry.
A key distinguishing feature of the various approaches mentioned above to create 3D hepatic spheroids is that they rely on scaffolds, but the aggregates are not themselves embedded in biomaterials, either naturally-derived or synthetic. While such biomaterial-free approaches can allow the cells to self-assemble and form their own ECM to surround the structure, ultimately they do not provide precise control over the structures, which may form differently across wells and experiments. Thus, several groups have investigated embedding hepatocytes either as single suspensions or as preaggregates into both natural and synthetic biomaterials. Of the many biomaterials being explored, hydrogels in particular have been adopted for 3D cell culture because their high water content and mechanical properties resemble those of native tissues. Furthermore, many hydrogels can be polymerized in the presence of cells, thereby ensuring a uniform cellular distribution throughout the 3D network. We refer the reader to a more comprehensive review of the various types of biomaterials that have been used for 3D culture of hepatocytes. 12 Here, we focus on polyethylene-glycol (PEG)–based hydrogels, pioneered by Lutolf and Hubbell. 131 PEG hydrogels are of great interest for cell culture due to their biocompatibility, hydrophilicity, and ability to be customized by varying chain length (to control microporosity and thus mechanical properties) or by chemically adding biological molecules that can allow cells to attach to the gel and/or modulate their phenotype ( Fig. 9 ). PEG hydrogels also allow for the incorporation of moieties that are sensitive to cell-secreted proteases, thereby allowing cellular remodeling of the gels. 132
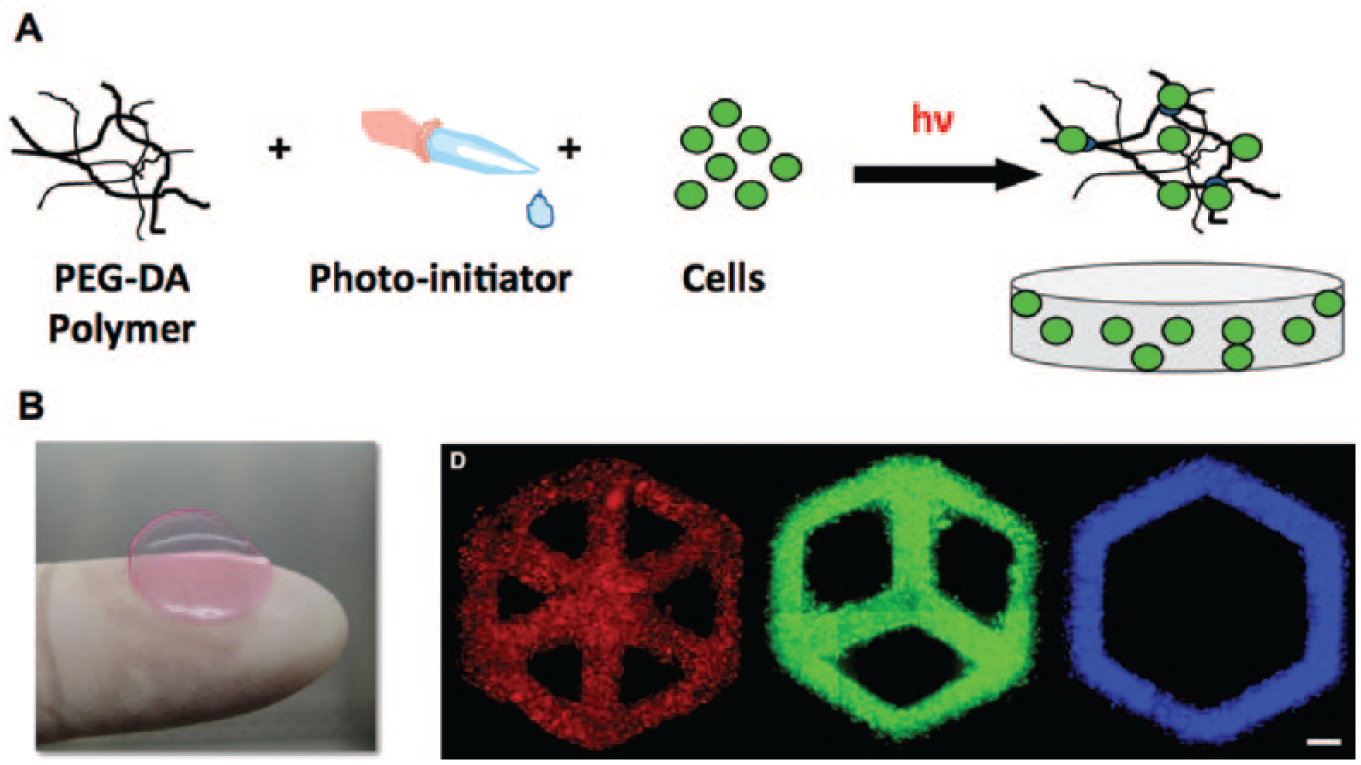
Creation of cell-laden hydrogels using polyethylene glycol (PEG). PEG-diacrylate (PEG-DA) is mixed with cells and photoinitiator and then exposed to UV light to cause gelation, thereby encapsulating cells (
In the case of the liver, the Bhatia group has cultured primary hepatocytes and supportive NPCs (fibroblasts, endothelia) in PEG hydrogels containing the RGD (arginine-glycine-aspartic acid) peptide to ligate hepatocyte surface integrins and enable long-term survival and functions.133,134 In addition, patterned photomasks have been used to localize the UV exposure of the prepolymer PEG solution and thereby dictate the structure of the resultant hydrogel. 133 More recent work showed that first stabilizing hepatocytes in the previously described 2D MPCCs and then lifting them off as “pucks” via collagenase for subsequent encapsulation in PEG hydrogels led to higher liver functions than cocultures that were randomly distributed in the hydrogels. 105 PEG microtissues generated using such pucks, coupled with a microfluidic droplet generator, were shown to be amenable to high-throughput drug-mediated CYP450 induction studies using a large particle flow cytometer.
Perfused Culture Systems
Bioreactors are devices in which the biological and/or biochemical processes develop under closely monitored and tightly controlled environmental parameters such as pH, temperature, pressure, nutrient supply, waste removal, and shear stress. A plethora of different bioreactor designs have been described in the literature for the culture of 2D and 3D liver tissue constructs for clinically relevant bioartificial liver devices.135–137 Here, we focus on small-scale bioreactors that have been developed for drug screening applications.
Physiological shear stresses play an important role in facilitating the phenotype of vascularized tissues under healthy and diseased conditions; however, the direct shear stresses experienced by hepatocytes are mitigated by their separation from sinusoidal blood by LSECs and the ECM-laden Space of Disse. The cell and ECM-mitigated reduction in shear is probably one of the reasons why static hepatocyte culture models (2D and 3D) have proven very useful for enabling long-term hepatic functions and for predictive drug screening as described in the previous sections. Nonetheless, liver perfusion introduces and removes molecules in the blood (i.e., oxygen, nutrients, hormones), which produce molecular gradients that modulate the hepatocyte phenotype in zones from the portal triad to the central vein (referred to as ‘zonation’). Simple gradients have been generated in vitro by progressive depletion of a substrate, such as oxygen, in a parallel-plate reactor, resulting in compartmentalized rat hepatic functions (i.e., CYP450 enzymes) as observed in vivo. 138 An experimentally validated mathematical model of oxygen transport within the parallel-plate bioreactor allowed for the prediction of oxygen gradients given the inlet partial pressure, cell-specific oxygen consumption rates, cell density, chamber length, media height, and flow rate. When a similar reactor was dosed with acetaminophen, increased toxicity was observed within regions of the hepatocyte monolayer that had higher expression of CYP450s due to exposure to lower tensions of oxygen (i.e., pericentral), as observed in liver sections from rats dosed with acetaminophen in vivo ( Fig. 10 ). 139
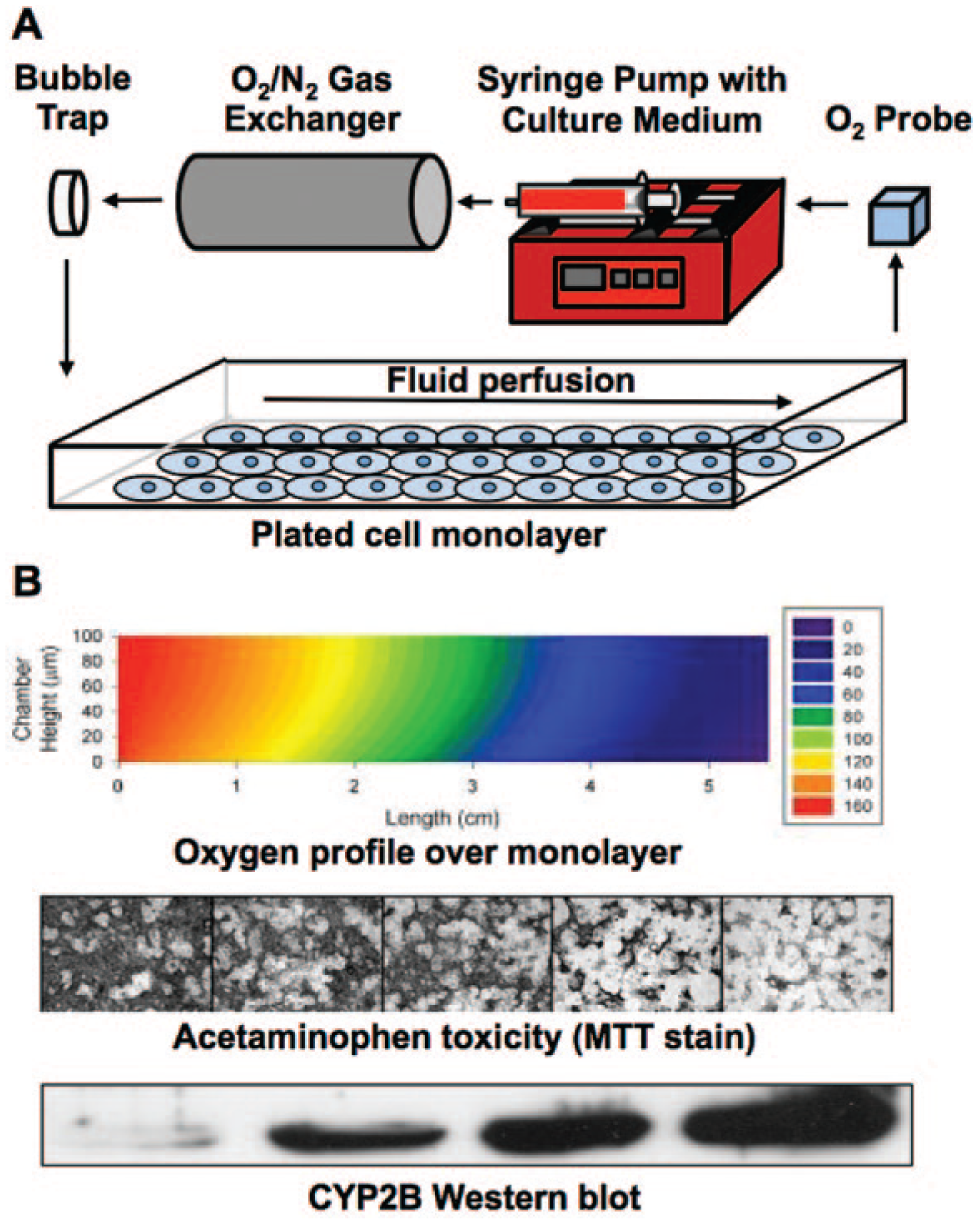
Parallel-plate bioreactor to expose cells to an oxygen gradient. Schematic of an experimental apparatus for a parallel-plate bioreactor is shown (
In addition to creating gradients of molecular factors, bioreactors facilitate automated delivery of nutrients and other soluble factors to cultures as well as the removal of waste products. In an era when an ever increasing array of chemicals are being produced in both the pharmaceutical and chemical industries, the automation and miniaturization advantages of microfluidic devices, especially those that can be parallelized for high-throughput screening, are likely going to play important roles in next-generation liver tissue devices. For instance, Eschbach et al. 140 designed a planar polymer scaffold with 900 microcontainers encompassing laser-drilled wells for perfused culture of 3D hepatic aggregates of fairly uniform sizes. In another platform by the Griffith group, an array of microchannels created via deep-reaction ion etching of silicon or polycarbonate wafers was used to culture preformed hepatic aggregates, which adhered to the collagen-coated walls of the etched channels ( Fig. 11 ). 123 A model of O2 consumption/transport in the culture medium was used to predict appropriate operating parameters for the cell cultures. Oxygen concentrations in the system were then measured as a function of flow rate and time after initiation of culture to determine the actual O2 consumption rates by the cultures. NPCs (i.e., endothelia, Kupffer macrophages) could be included in these cellular structures. For example, endothelia self-sorted to the outer margins of the spheroids, which allowed for perfusion with flow rates similar to those observed in liver sinusoids. A higher throughput version with 12 bioreactors on a single plate has also been implemented. Functional results with this system demonstrated better retention of hepatic gene expression than conventional collagen sandwich cultures. Moreover, higher cytokine production was observed when the bioreactors were stimulated with LPS in the presence of Kupffer macrophages compared with control bioreactors containing only hepatocytes. Further assessment of metabolic clearance showed good correlation with human in vivo clearance data. 95 Tostões et al. 141 recently used an automated perfusion bioreactor to maintain key functions and gene expression of PHH spheroids for a few weeks in culture, thereby providing an avenue to conduct repeated drug dosing.
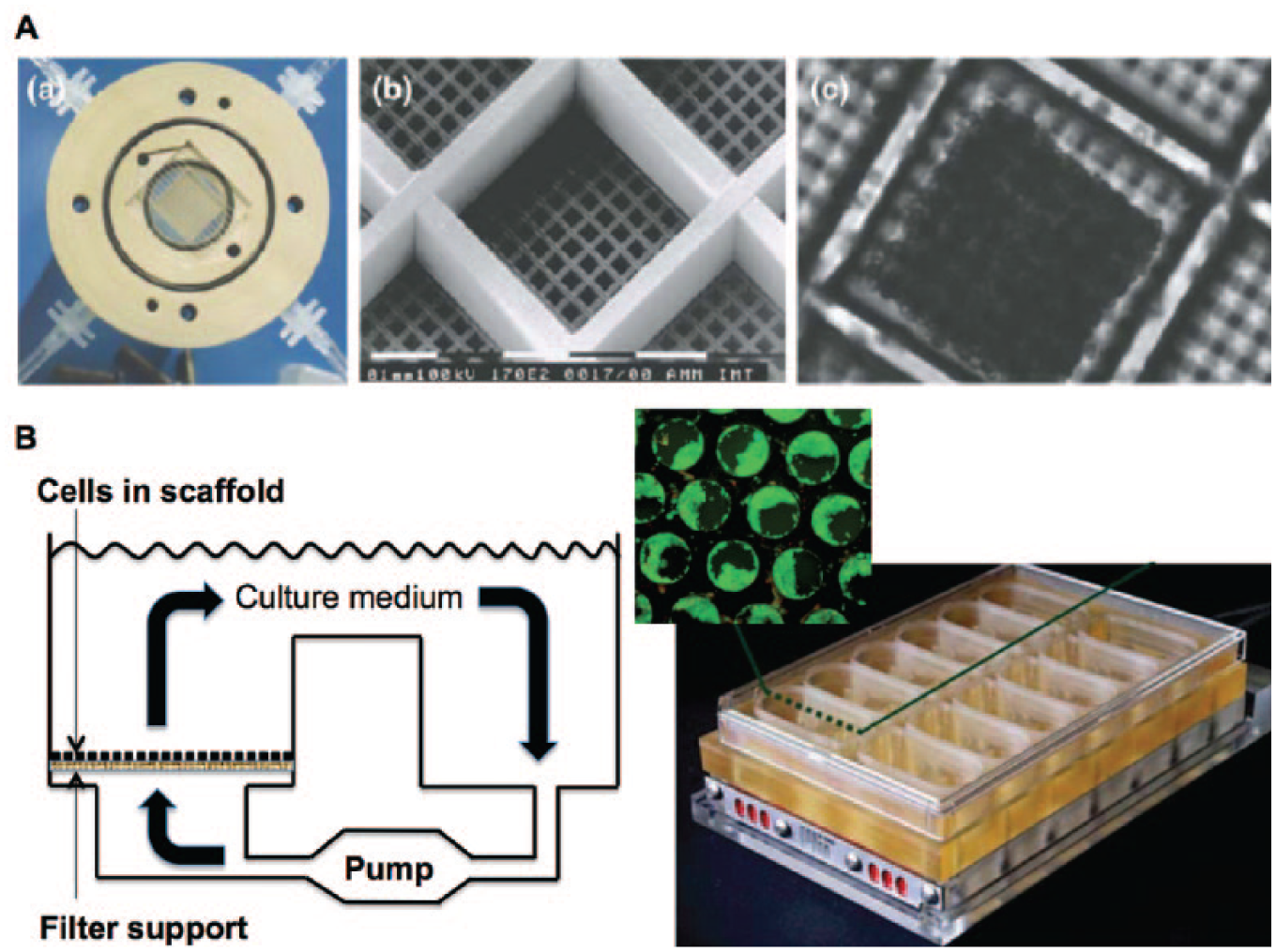
Bioreactors for culture of 3D hepatic spheroids. Planar polymer scaffold with microcontainers for the culture of uniformly sized 3D cellular aggregates (primary rat hepatocytes shown) (
Microfluidic structures, designed to mimic the liver acinus, have been used to form high-density 3D hepatocyte aggregates in channels that have through-holes, which allow for perfusion-based medium exchange through adjacent compartments. 142 These channels are subjected to gravity-driven flow in a 96-well plate footprint (32 independent flow units per plate), eliminating the need for external pumps and connections ( Fig. 12 ). A similar device design, using a micropillar array to allow diffusion of soluble factors to hepatocyte aggregates, was implemented by Toh et al. 143 (3D HepaTox Chip). Incorporated concentration gradient generators coupled with eight cell culture channels allowed a dose response for a given chemical to be obtained on the same chip. Goral et al. 144 devised a microfluidic device that promotes the 3D organization of hepatocytes into cord-like structures (as in vivo) without the addition of biological or synthetic matrices. In contrast, Liu Tsang et al. 133 subjected 3D hepatic tissues, which were created by photopatterning PEG-encapsulated hepatocytes, to perfusion in a bioreactor to improve liver functions.
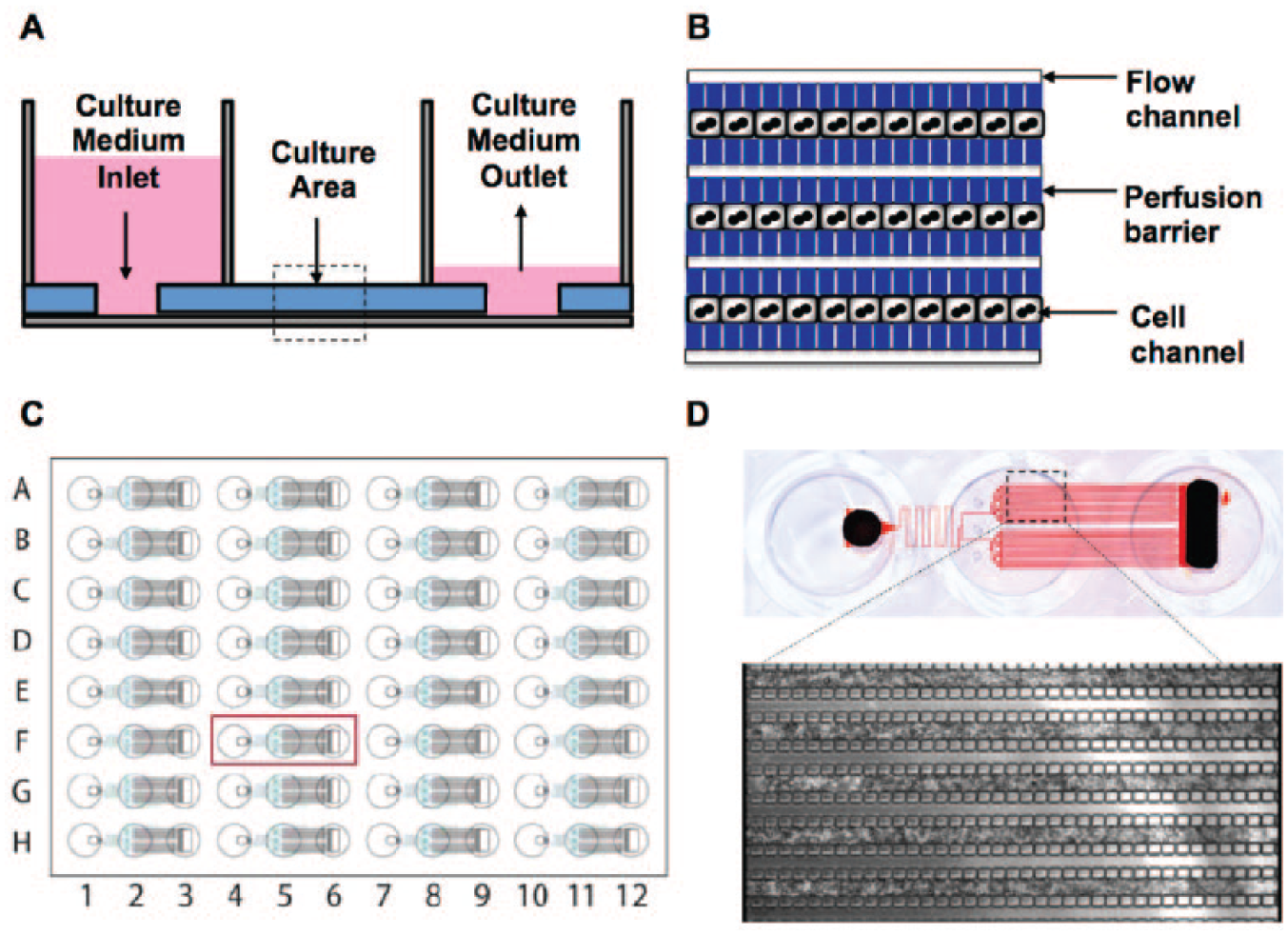
Microfluidic device for culture of hepatic aggregates. Gravity-driven flow allows perfusion through the aggregates (
Several groups have adapted the classic sandwich format or related variations to protect hepatocytes from shear stresses induced by direct exposure to flowing fluid. Xia et al. 145 have designed a flow bioreactor out of acrylic and an oxygen-permeable, collagen-coated porous membrane to protect rat hepatocytes from the impact of fluid flow. The authors evaluated the velocity profile and the mean fluid shear stress in their bioreactor using a computational fluid dynamics model to simulate flow and numerically solve the steady-state Navier-Stokes equations. Over 5 days of culture, the bioreactor with perfusion and oxygenation had significantly higher albumin production than the static control or the perfused bioreactor without oxygenation. Other bioreactors have been treated with acetaminophen and were shown to be more sensitive to toxicity than a static culture control. More recently, Hegde et al. 146 designed a microfluidic device with a porous membrane sandwiched between two chambers of PDMS. In the bottom chamber, hepatocytes are cultured in a classic collagen sandwich configuration, while the top chamber is used for culture medium perfusion. The authors observed higher albumin secretion, urea synthesis, CYP1A1 activity, and a more extensive bile canaliculi network in hepatocyte cultures that were subjected to flow over static controls. Perhaps more important, the authors showed that collagen synthesis was enhanced in flow cultures relative to static controls and that inhibiting collagen secretion reduced albumin production and created gaps in the canalicular network. Dash et al. 147 adapted a commercially available (HemoShear, Charlottesville, VA) cone and plate technology (previously used for subjecting smooth muscle cell and endothelial cell cocultures to physiological hemodynamics to restore vascular phenotype) to rat hepatocytes cultured in the aforementioned collagen gel sandwich. Albumin, urea, and CYP450 activities were improved over 2 weeks in cultures subjected to controlled hemodynamics over static control cultures. Importantly, CYP1A and CYP3A were inducible in the cultures via prototypical drugs at concentrations closer to in vivo plasma levels documented in rats.
Given the high oxygen uptake rate of hepatocytes, it is likely that perfusion in these platforms helps to limit necrosis in the core of cellular aggregates. Yet, even in 2D monolayers directly exposed to flowing fluid, Novik et al. 148 showed that flow-based cocultures of PHHs and endothelial cells were able to better predict in vivo drug clearance and produce drug metabolites at greater rates than static mono- or coculture controls. The authors suggest hypotheses around waste removal on top of cells and shear stress-induced cellular uptake of drugs to explain why flow-based cultures performed better than static controls for drug turnover. However, further studies are needed to test these hypotheses. Nonetheless, this example shows that perfusion-based hepatocyte systems may also be beneficial for 2D culture. In another example, Kane et al. 149 developed an 8 × 8 element nonaddressable array of microfluidic wells capable of supporting micropatterned hepatocyte-fibroblast cocultures. Two microfluidic networks independently perfuse the cocultures with culture medium and oxygen. Overall, throughput in flow-based systems is typically reduced over testing in static multiwell formats (i.e., 96-well plates); however, further on-chip miniaturization and automated control of fluid flow within engineered tissues are rapidly evolving to meet the throughput needs of the pharmaceutical industry.150–152
Perfusion is also necessary for the creation of organs-on-a-chip platforms, where different tissue compartments can interact with each other through the sharing of secreted molecules (i.e., drug metabolites, hormones). Such chips are especially useful to model the pharmacokinetics and pharmacodynamics of drugs in a single device. After initial work by Viravaidya et al., 153 the concept of organs-on-a-chip has recently been touted as the next frontier in drug screening and disease modeling, bringing us closer to predicting in vivo outcomes across not just the liver but multiple organs interacting as a system. Recently, several groups, backed by major funding initiatives from NIH and DARPA, have embarked on the creation of modular organs-on-a-chip using both primary hepatocytes and iHeps. We will refer the reader to other reviews that discuss the design principles behind organs-on-a-chip systems.150–152,154 Here, it suffices to state that each tissue compartment in an organs-on-a-chip system needs to be optimized for in vivo–like functionality and then tested in a fully integrated device with interconnections between multiple tissue types. The use of suboptimal tissue models (i.e., hepatic cell lines, declining PHH cultures), even if connected via flow, is likely not going to advance the field forward over what has already been accomplished in this arena. Finally, given the staggered advances in culture systems for different organs, creation of modular microfluidic designs that can accommodate a “plug-and-play” approach to incorporating different tissue models will likely be necessary for rapid development and validation of evolving versions of organs-on-a-chip platforms.
Applications of Engineered Liver Models in Drug Development
In vitro models of the liver are often used throughout drug development for applications in drug disposition and drug-drug interactions (DDIs).106,155,156 In particular, microsomes are used to evaluate which CYP450 enzymes metabolize a given class of compounds, suspension hepatocytes and some plated formats are used to predict clearance rates of compounds, plated hepatocytes are used to evaluate induction of key CYP450 enzymes to predict DDI potential, and plated cultures from specific “qualified” hepatocyte lots are used for evaluating transporter-drug interactions that can affect overall drug disposition. Hepatocytes from animal species (i.e., rat, dog, monkey) are also used alongside human liver models for in vivo species selection for efficacy and toxicology as discussed earlier. However, one major issue with the use of different liver models for the aforementioned applications is that it makes data integration and interpretation nontrivial. For instance, if a drug inhibits a certain CYP450 at a given concentration in microsomes, such may not be the case in plated PHHs that may actively pump the drug out of the cytoplasm via transporters. Furthermore, in vitro liver models are not as widely used for routine drug toxicity screening as they are for drug disposition and DDI investigations, since the sensitivity for prediction of clinical toxicity outcomes can vary considerably from one model to another and is typically low in conventional culture models (<50%).
Most vendors of commercially available PHHs have adopted the strategy of qualifying the lots as suitable for suspension or plated culture and then, within plated cultures, lots that are suitable for metabolism, induction, or transporter studies. Criteria for such qualifications can vary across vendors but typically are based on quantitative thresholds of cell responses (i.e., fold changes in enzyme inductions) following incubation with prototypical drugs. Furthermore, given the variability across hepatocyte lots in functions, the idea of pooling many donors into a single lot has become common practice to model an “average” human liver. Use of pooled lots for plated cultures can be problematic, however, given differences across donor hepatocytes in attachment efficiency to ECM-coated surfaces. Such upfront qualifications and pooling can drive up the cost of the hepatocyte lots given the added studies required.
When cultured in a more sophisticated and stabilizing platform (i.e., micropatterned cocultures), PHHs have a tremendous capacity to recover and become highly functional.74,106,107 In addition, any quality issues related to the trauma of isolation, cryopreservation, and thawing tend to dissipate with prolonged culture, at least for a majority of the PHH lots in our experience.24,157 Thus, while engineered liver models provide for higher prediction of clinical outcomes than conventional models, they also allow PHHs from different donors to recover in vitro to be then used for acute (days) or chronic (weeks) drug dosings. If functional assays are nondestructive (i.e., enzyme induction or albumin secretion), long-term engineered liver models can allow for reuse of the cultures in many cases, while using fewer seeded PHHs than traditional confluent monolayers. All of these advantages help mitigate batch-to-batch quality issues across PHH lots, bring out the true differences in drug responses due to each donor as opposed to differential trauma of isolation, and allow for the creation and use of a greater number of engineered devices using valuable PHH lots. Below, we discuss applications of PHH-based liver models in further detail.
Drug Metabolism
Prediction of drug clearance and identification of major drug metabolites are key applications of in vitro liver models.106,156,157 Clearance prediction allows for proper dose selection in animal studies and human clinical trials, while identification of major metabolites (greater than or equal to 10% of drug-related material158,159) allows for the assessment of metabolite efficacy and safety in preclinical testing prior to initiation of human clinical trials. However, it is now well established that most lots of suspension PHHs, especially those derived from single donors, have very limited ability to predict the clearance of compounds that have low turnover (i.e., once-a-day dose) in vivo. 157 In most cases, the compound does not turn over at all, failing to allow any clearance prediction. The pharmaceutical industry is now developing many low-turnover compounds, as once-a-day dosing regimens are likely better for the end user’s ability to comply with medication instructions. Therefore, using human liver models to obtain accurate clearance predictions is of considerable interest, especially if there is lack of concordance between animals and humans in metabolism for a given class of compounds. The relay method developed by Pfizer (New York, NY) now allows suspension PHH lots, pooled from many donors, to be used sequentially (i.e., drug incubated across multiple thawed cell vials) to obtain enough turnover of low-clearance compounds to be able to predict in vivo clearance. 160 However, selection of specific pooled cryopreserved PHH lots and banking for future use are important for this method, as not every lot will work well with this strategy.
Cocultures of hepatocytes and supportive NPCs have proven useful for predicting clearance of compounds with a wider range of in vivo turnover rates, including low-clearance compounds.148,157 In our experience, cocultures allow for the use of single donors to accomplish this goal without the need to select for the optimal pooled lot, as in the aforementioned relay method. Regardless, based on recently published studies,157,160,161 it appears that while there are some pending issues as to how best to model drug-protein binding in vitro for more accurate clearance prediction, the previously intractable problem of clearance prediction for low-turnover compounds is now being implemented during the drug development pipeline.
The identification of major human-relevant drug metabolites in vitro is fraught with problems due to missing enzymes and transporters in cell-free microsomes, the inability to incubate for more than a few hours in suspension PHHs (at least with a single thawing), and the severely reduced metabolic capacity of conventionally plated PHHs compared with the native liver. For such reasons, ~50% of clinically relevant major drug metabolites are missed in conventional model systems.106,162 For secondary metabolites, which are typically several reactions from the parent molecule and may take some time to generate, the miss rate increases to ~62% (38% identification rate). When the same compounds are used in the MPCC model system created using PHHs, the identification rate increases to 75% and 67% for total and secondary metabolite generation, respectively. 106 Increased identification rates are partly due to the higher per hepatocyte enzymatic activities in MPCCs and the ability to incubate for up to 1 week without a medium change to identify slowly generated secondary metabolites. Higher production of drug metabolites has also been observed in other engineered liver models.123,148 The contribution of the stromal compartment to metabolite generation also has to be considered by dosing of stromal-only control cultures with the chosen drugs. Some metabolites are still missed in these engineered liver models, potentially due to very low expression of certain enzymes in current models and extrahepatic metabolism, either in liver NPCs and/or in organs other than the liver (i.e., intestine). Continued improvements in liver culture systems, including their incorporation into organs-on-a-chip systems, should help address such deficiencies.
Transporter Interactions
Both uptake and efflux transporters, distributed over many organs, are now recognized as major contributors to overall drug disposition in the body.163,164 Thus, a variety of in vitro models have been developed to assess transporter contributions. In the case of hepatocytes, one of the major limitations of existing systems is that the intrahepatic bile canaliculi are typically not completely formed, and the canalicular network is not connected to a biliary tree as in vivo. Furthermore, in our experience, the canaliculi are “leaky” in cultured hepatocytes, and the canalicular contents end up being present in the culture medium, which makes it difficult to determine whether a drug and/or its metabolites are going to be excreted into the blood or the bile compartment. Despite these limitations, Ghibellini et al. 165 used buffers with or without calcium to get a measure of drugs that are excreted into the canaliculi. Briefly, one set of cultures is incubated with drug in the presence of calcium, which is required to maintain the integrity of tight junctions holding the canaliculi together. Another set of cultures is incubated with the same drug but in calcium-free buffer, which causes all the bile canaliculi to be disrupted and any material excreted into the canaliculi to almost immediately show up in the culture medium without any resident time in the canalicular network. Evaluating the drug contents in the cell lysates in these two incubations, using either liquid chromatography/tandem mass spectrometry (LC-MS/MS) or radiolabeled drugs, allows the investigator to get a measure of how much drug-related material was excreted and then retained transiently in the bile canaliculi. This technique has been adapted to PHHs for use during drug development. 166
The technique established by Ghibellini et al. 165 has been attempted, for the most part, with hepatocytes cultured in the ECM-sandwich format. While this format allows robust networks of bile canaliculi to form in 4 to 6 days, the CYP450 enzyme activities are downregulated significantly by that time in culture.11,74 Therefore, the model is limited to evaluation of parent drug efflux into the canaliculi. To evaluate the interplay between drug metabolism and transport, an engineered culture model that can form both the bile canaliculi and maintain high levels of enzymatic activities may be more suitable. Ultimately, however, separate biliary and blood compartments will be needed in engineered culture models to evaluate the composition of drug-related excretions directly. No current system can provide both compartments, although some designs have been proposed. 167 It is possible that coculture with cholangiocytes (cells that form small biliary ductules) in vitro may provide clues as to how these cells interface with hepatic bile canaliculi to drain the biliary contents in vivo. Furthermore, transporters are ubiquitous in multiple organs of the body, and understanding how they affect the overall clearance of a given drug from the body will be important in the future. Organs-on-a-chip systems may find utility in addressing such questions.
Drug-Drug Interactions
Administration of one drug (perpetrator) can affect the efficacy and/or toxicity of a coadministered drug (victim), resulting in DDIs. There have been several cases of DDIs in the clinic, and now warnings on drug labels for such interactions are common.168,169 In many cases, DDIs occur via drug-mediated induction or inhibition of CYP450 enzyme activities. Nuclear receptors play important roles in drug-mediated CYP450 induction, while inhibition can be competitive or time-dependent. The FDA has put forth a guidance document that lists the types of in vitro studies with human-relevant systems that can be performed to assess the potential for DDIs and allow companies to develop clinical trials to further probe the risks. 169 This is one area where conventional cultures of PHHs have made a tremendous impact. For instance, typically three donors of PHH cultures are used for 1- to 3-day incubation periods with a new drug candidate of interest. Transcript and activity levels of CYP1A2, CYP2B6, and CYP3A4 are measured, as these enzymes are regulated by the three major nuclear receptors, AhR (aryl hydrocarbon receptor), PXR (pregnane X receptor), and CAR (constitutive androstane receptor). The candidate drug-mediated increases in transcript and enzyme activities relative to the vehicle control (typically fold changes) are benchmarked to increases observed with prototypical inducer drugs that are known to cause clinical DDIs. Overall, mRNA transcript assessment has been shown to be a more sensitive measure of induction, rather than enzyme activity. 170
While the aforesaid protocols for use of PHHs in predicting DDIs constitute an advance for in vitro human-relevant liver models during drug development, there are some key limitations. First, the fold induction levels that are obtained with declining PHH cultures can be unusually high (>50 fold) due to the very low baseline (nondrug) enzyme activities. While such high fold changes provide a higher dynamic range for rank ordering drugs of interest, they do not always predict the fold induction levels observed in the clinic. Second, because PHH cultures often have very low enzyme activities, the role of metabolites in causing enzyme induction and inhibition cannot always be evaluated effectively. Third, DDIs at the level of transporter inhibition cannot be fully appraised given the low expression and mislocalization of some key transporters on apical and basolateral membranes of hepatocytes in conventional culture systems. Fourth, drug effects on enzyme induction/inhibition over several weeks (as can occur in the clinic) cannot be fully appraised in short-term conventional formats. Finally, while enzyme induction/inhibition and transporter interactions underlie DDIs, ultimately the clinical concerns are related to reduction in efficacy (i.e., due to increased drug clearance) or increase in toxicity (i.e., due to reduced drug clearance or production of toxic drug metabolites) of victim drugs following administration of perpetrator drugs. The effects of enzyme induction/inhibition on drug efficacy and toxicity cannot be easily tested with short-term cultures given the length of time required to first induce CYP450s in the cultures (~3 days) and then evaluate toxicity with chronic dosing regimens (>1 week). Engineered liver models that possess high levels of enzyme activities and nuclear receptor activity for a few weeks can potentially address the aforementioned issues. Nonetheless, enzyme induction studies in PHHs are now appearing in FDA guidance documents, demonstrating that in vitro human liver models can indeed make an important impact in the drug development pipeline. We anticipate that future FDA guidance documents on other applications will also include the use of human-relevant in vitro liver models.
Infectious Diseases
Pathogens that infect the liver are of serious global concern. For instance, hepatitis C virus (HCV) and hepatitis B virus (HBV) chronically infect the livers of 130 to 170 million and 400 million people worldwide, respectively. Moreover, the Plasmodium protozoan, infecting over 250 million individuals with malaria, matures within the liver. While clinical management of these diseases has improved considerably in recent years, there is considerable room for improvement. For instance, prophylactic options for HCV are not available, and current therapies have serious side effects as well as high costs. For malaria, limited prophylaxis is available, and only a few drugs currently target liver-stage parasites. However, drug resistance remains a growing problem, and only one licensed drug eliminates the dormant hypnozoite form of the pathogen that is responsible for clinical relapses. For HBV, lifelong treatment is often required because of the stable nature of viral episomal DNA (covalently closed circular DNA), which maintains basal levels in infected cell nuclei, even upon nucleoside/nucleotide inhibitor treatment. An improved understanding of the pathogenesis of these hepatic diseases within human hosts will likely help create better clinical therapies. Toward that end, robust infectious model systems must be designed to support both productive viral infection and accurately mimic virus-host interactions. Furthermore, such systems should be able to model the interplay between drug metabolism, toxicity, and efficacy, as all three play an integrated role in ultimately determining the effectiveness of a given drug treatment.
As mentioned earlier, animal models can be expensive (i.e., chimpanzees for studying HCV infection), are lower throughput, and can raise ethical concerns. Although cancerous and immortalized cell lines are capable of supporting the entire life cycle of some of these pathogens, they have significant abnormalities in liver functions (i.e., uncontrolled proliferation, dysregulated gene expression, altered host responses to infection, inadequate drug metabolism capacity, dysfunctional mitochondria, abnormal endocytic functions), which inhibits the accurate study of host-virus interactions.8,35 Furthermore, established cell lines ultimately only reflect the pathophysiology from one human donor. Therefore, PHHs are considered the gold standard for studying these pathogens.
Conventional culture formats have been shown to support long-term infection of PHHs with the aforementioned pathogens.171–178 However, PHH phenotype declines rapidly for most donor lots, making routine implementation of such models for drug screening very difficult for infectious diseases. Furthermore, in our experience, not all PHH lots support long-term infection in conventional culture models. Major advances have been made by the Bhatia group in using MPCCs of stable PHHs and 3T3-J2 fibroblasts to study the infection and drug response for HCV, HBV, and malaria, as well as discover novel infection biology ( Fig. 13 ).108–110 Interestingly, both conventional monolayer cultures and randomly distributed cocultures of the same two cell types were not able to sustain infection of any of the pathogens above, suggesting that stability of hepatic phenotype and proper hepatocyte polarity, as mediated by the control over cell-cell interactions (i.e., architecture), are critical for enabling long-term infection in multiple PHH donors. For HCV and HBV, the Bhatia group has also shown infection in cultures of iHeps, opening up avenues for studying the effects of donor genotype and host genes on infection efficiency, propagation, and resistance to drug therapies.110,179
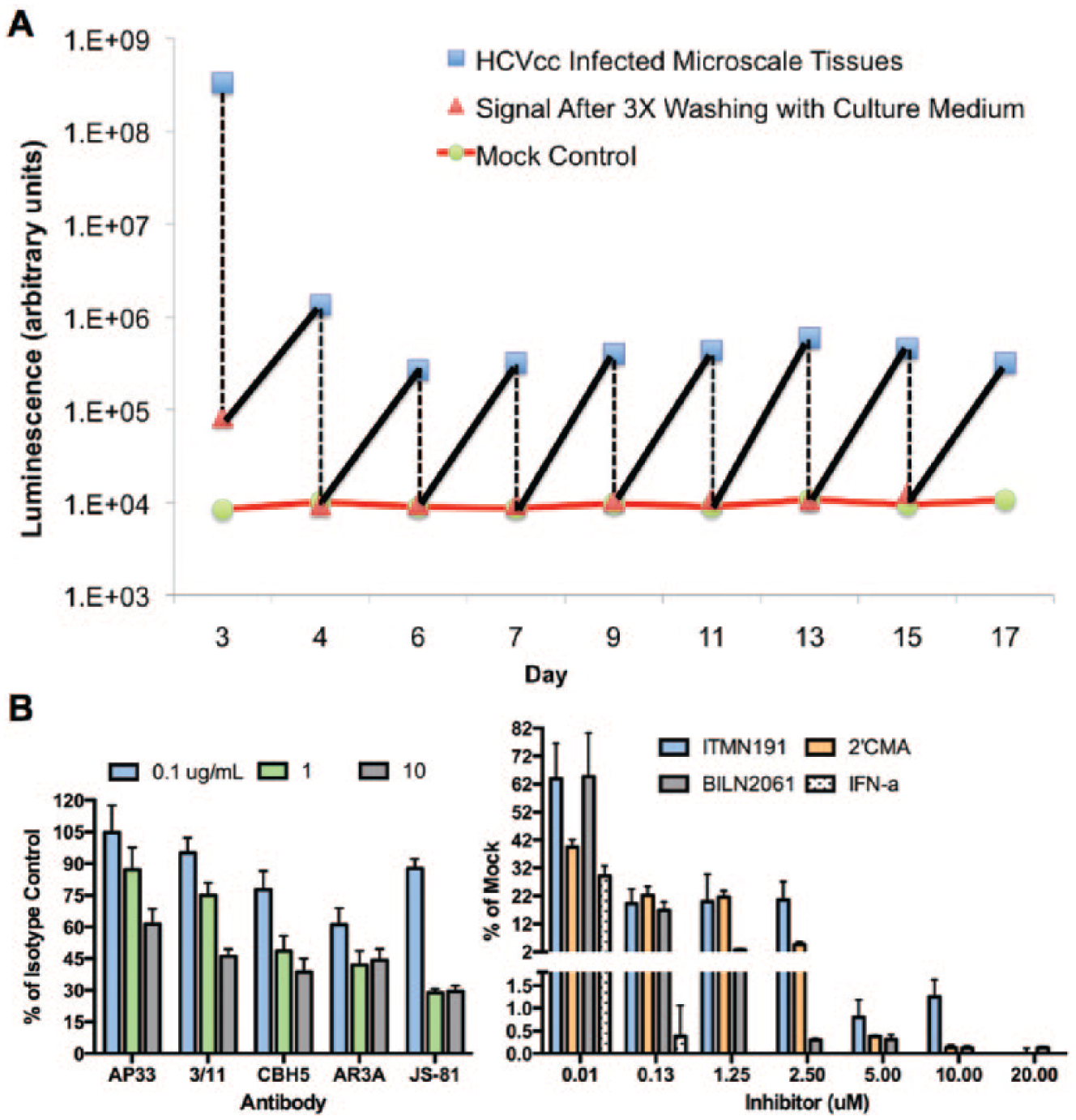
Hepatitis C virus (HCV) infection in engineered cultures of primary human hepatocytes (PHHs). Micropatterned cocultures (MPCCs) of PHHs and 3T3-J2 murine embryonic fibroblasts in a 96-well format were infected for 24 h with cell culture–adapted HCV with a secreted luciferase reporter (i.e., HCVcc). Cultures displayed stable infection for several weeks in vitro. The sawtooth pattern on the graph is a result of culture medium changes every 2 days followed by viral replication and secretion of luciferase in supernatants (
Despite impressive progress in the use of engineered MPCCs to enable sustained pathogen replication, the infection efficiency remains very low (i.e., <5% for HCV). While some PHH lots display stable functions in MPCCs for several weeks, they remain refractory to infection with one or more of these pathogens. Such issues limit the use of multidonor MPCCs for routine drug screening applications in infectious diseases. These examples also demonstrate two important principles in interfacing engineered systems with PHHs for infectious disease applications: (1) robust liver phenotype over several weeks is a prerequisite for infection of PHHs with pathogens, and (2) selection of a donor lot is critically important and requires upfront validation studies with a specific pathogen of interest. Better understanding of host-virus interactions will be necessary to cut down on the time required for donor selection and/or devise strategies to allow more PHH lots to be used for a given application. One interesting example is the use of a broad-spectrum Janus kinase (JAK) inhibitor to attenuate the innate immune response in PHHs, which allowed them to take up HCV and HBV more efficiently than in cultures not treated with the inhibitor. 110
Drug-Induced Liver Injury
Drug-induced liver injury (DILI) is a leading cause of both the prelaunch and postmarket attrition of pharmaceuticals. 4 There are three significant issues underlying why DILI is often missed in preclinical drug testing. First, in vitro models of the human liver used by pharmaceutical companies, while having good specificity (i.e., low false-positive rates <5%), are only between 30% and 50% sensitive in classifying diverse classes of drugs as liver toxins.8,180 Low sensitivities in toxicity classification are partly due to the inability to dose drugs chronically over weeks to months, culture adaptions resulting in very low levels of enzymatic activities in hepatocytes, and missing physiologic interactions with liver NPCs and the adaptive immune system. Second, even when in vitro models classify drugs as hepatotoxic, the doses needed to elicit toxicity are considerably higher than the concentration in blood at which a therapeutic effect could be attained. Indeed, in a landmark study by Xu et al., 180 doses up to 100-fold of Cmax (maximum concentration of drug in human blood) were required for many drugs to produce an adverse effect in collagen/Matrigel sandwich PHH cultures. The authors justify the use of such high doses because different individuals often have variable drug concentrations in their blood due to polymorphisms in drug metabolism enzymes and other genes, and the blood concentration of a drug may not necessarily reflect the drug accumulation in the liver due to the presence of transporters. However, the use of very high drug doses could also partly be necessary due to much lower enzyme activities in cultured PHHs compared with in vivo levels, as well as the inability to dose sandwich cultures for several weeks or months to cause accumulation of toxic drug-related material (i.e., metabolites), as could be the case in the clinic for drugs being administered to treat chronic diseases in patients. Third, while there may be drug-induced hepatic dysfunction or even death in vitro, how the hepatocytes in the liver of a specific individual may adapt to initial injury is something that cannot be easily predicted using current approaches with a few lots of PHHs. For all these reasons, pharmaceutical scientists often will rely on tests in live animals to get a measure of whole-liver and whole-body drug effects. Yet, significant differences between animals and humans in liver functions lead to lack of concordance in drug responses for several drug classes. 6
The drawbacks with current in vitro models and live animal studies are key motivations underlying the development of more physiologically relevant in vitro models of the human liver. We have shown that when MPCCs created with PHHs were subjected to repeat drug doses over 9 days (4 total dose administrations), the sensitivity of drug toxicity detection improved to 70% to 75% (without compromising specificity of the assay) compared to ~30% sensitivity in sandwich cultures created from the same PHH donor and dosed for 24 h with the same drug set (35 toxins, 10 nontoxins). 107 The higher sensitivity of MPCCs is partly due to their ability to generate greater amounts of toxic drug metabolites (i.e., for acetaminophen) as well as the accumulation of such metabolites over 9 days of dosing. Analogue compounds with differential clinical toxicity were also picked up correctly in MPCCs, paving the way for use in prospective drug screening campaigns to prioritize structural analogues, even in the absence of human clinical data (i.e., Cmax). Furthermore, rat hepatocytes in MPCCs were only ~50% sensitive with the same set of drugs and dosing regimen, thereby showing species-specific differences in hepatic sensitivity to drug toxicity in the same culture platform.
Our drug toxicity data mentioned above have confirmed studies by others showing that some idiosyncratic toxins (i.e., zafirlukast, troglitazone) can be detected using cellular stress markers in vitro,8,180,181 potentially because hepatic stress is often a first step in the cascade of mechanisms that cause overt liver injury in specific patients with one or more covarying genetic (i.e., P450 polymorphisms) and environmental (i.e., coadministered drugs, alcohol) factors. However, due to the limited availability of PHH donors for use in MPCCs (and other current liver models for that matter), it is not yet possible to determine why some livers may recover from the initial hepatic stresses, whereas others may show rapid DILI progression. Panels of iHeps with specific genetic polymorphisms coupled with the ability to superimpose environmental factors in long-term, multicellular culture platforms may provide an avenue for evaluating patient-specific liver adaptation following cellular stresses. Nonetheless, our data and that of others show that engineered liver models can provide a sufficiently sensitive readout, at least for an initial drug toxicity screen, to prioritize compounds from the same class for further development. If such a screen is done early enough in drug development (i.e., drug discovery), medicinal chemistry can be leveraged to develop less toxic structural analogues based on structure-activity relationships (SARs).
High-Content Readouts for Prediction of Drug Toxicity
An important line of research is the development of high-content screening (HCS) fluorescent readouts to provide a mechanistic understanding of how drugs cause hepatic stresses. Several HCS systems have been commercialized, coupling automated and multispectral epifluorescent microscopy, software for real-time analysis of fluorescent intensities within individual cells, and databases for storage of the massive amount of individual cell data generated from multiwell plates (i.e., Thermo-Fisher ArrayScan, Molecular Devices ImageXpress, GE Healthcare IN Cell Analyzer). Fluorescent probes for key organelles/events within cells (i.e., mitochondria, lipids, reactive oxygen species, nuclei) are also commercially available to interface with HCS instruments. Evaluation of hepatotoxicity using multiparameter cell feature analysis measured by fluorescent imaging was implemented by O’Brien et al. 181 using HepG2 and was later extended by Xu et al. 180 to short-term ECM sandwich cultures of PHHs. More recently, HCS has been adapted to monolayers of iHeps, although comparisons to PHHs were lacking. 182 CellCiphr Profiling by Cellumen (now offered by Cyprotex, Watertown, MA) was the first commercial assay introduced in 2007 to predict hepatotoxicity using multiparameter HCS-based fluorescent probe measurements. A predictive risk assessment is calculated in this assay using a classifier model comparing the in vitro cell signatures (in hepatoma cell lines and rat hepatocytes) against animal preclinical toxicity data.
HCS approaches have recently been adapted to micropatterned cocultures of PHHs and 3T3-J2 fibroblasts.62,115 The development of computational algorithms, designed to separate out the fluorescent intensities from multiple cell types in cocultures, has now set the precedence for using HCS in more sophisticated multicellular models in the future. Whether HCS readouts are more sensitive (i.e., lower IC50 values) for identifying toxic compounds over more standard end points (i.e., adenosine triphosphate [ATP], albumin, urea) has not been determined in the same study with the same drug set using stable cultures of PHHs (as opposed to declining ones). Regardless, multifluorescent imaging of key organelles provides important information about a drug’s mechanism of toxic action in liver cells, as chronic organelle dysfunction has been implicated in the liver’s inability to adapt in certain cases of DILI.180,183,184
Toxicogenomics (TGx) combines genomics (i.e., mRNA transcripts, microRNAs, DNA methylation patterns, single-nucleotide polymorphisms) and bioinformatics analyses to identify and characterize the mechanisms of action for suspected toxicants. 185 One goal of TGx is to identify sets of genes that may be candidate biomarkers of specific toxic effects. These biomarkers may subsequently serve to classify drugs as potentially toxic in early stages of drug development and/or provide early detection signals in clinical settings prior to full-blown onset of liver enzyme elevations. The majority of TGx studies have been performed in rats.185,186 However, while the proof of concept has been established for the value of TGx in preclinical drug development, the sensitivity of DILI predictions between rats and humans has not been adequate, presumably due to the species-specific differences in drug metabolism pathways. For instance, an FDA study found that the DILI potential of a drug on humans can only be reasonably assessed using TGx analyses of in vivo studies in rats if the drug produced significant elevation of alanine aminotransferase (ALT) or total bilirubin (TBL). 187 But when ALT or TBL levels were not elevated in vivo, the TGx approach was not sufficiently robust to predict human outcomes. Similarly, a consortium of pharmaceutical companies and universities in Europe carried out studies in rats dosed with 16 compounds over 2 weeks. They concluded that the combined approach of “omics” was a very useful tool for the generation of mechanistic hypotheses but only in conjunction with conventional toxicology readouts. Furthermore, proteomics and metabolomics were limited to being supportive of the findings at the transcriptomic level. 188 Thus, the use of tissue transcriptomics still needs to be coupled with histopathology to deliver the best results.
Cultures of PHHs can potentially address the limitations associated with using TGx analyses for those drugs that do not cause overt liver enzyme elevations in rats. However, while TGx studies in PHHs have shown some benefit in identifying liver pathways affected for a few drugs, progress has been stymied due to the “destabilization” (dedifferentiation) of the gene expression/phenotype that occurs in these cells in conventional culture formats. 189 Indeed, one study found significant gene expression changes when hepatocytes were cultured on collagen as opposed to Matrigel, thereby showing that the culture platform is an important consideration for TGx studies using hepatocytes. 190 Functionally stable engineered liver models may address such shortcomings in using TGx more effectively during preclinical drug development.
In Silico Prediction of Drug Effects
To evaluate the predictive signatures generated by any liver model, clinical and detailed mechanistic information on the hepatic effects of large compound sets is required. The FDA has been compiling the Liver Toxicity Knowledge Base (LTKB), which collects diverse types of data on individual marketed drugs, such as mechanisms of liver injury, histopathology, drug metabolism, and side effects. 191 The NIH has also established the LiverTox website (LiverTox.nih.gov) to provide comprehensive toxicity data and extensive references on drugs, herbal supplements, and dietary supplements. A commercially available database, PharmaPendium, provides preclinical, clinical, and postrelease safety data on FDA-approved drugs. ACToR (Aggregated Computational Toxicology Resource) is a database software that contains information on chemical structures, in vitro bioassays, and in vivo toxicology assays derived from >150 sources such as the U.S. Environmental Protection Agency (EPA), Centers for Disease Control and Prevention (CDC), the World Health Organization (WHO), and the FDA. 192
The information stored in current databases will need to be integrated with cellular functions and molecular markers (i.e., those generated from high-content screens as described earlier) to better enable in silico predictive modeling. For instance, the ToxCast project by the EPA is evaluating diverse in vitro assays for understanding different types of molecular and pathway perturbations caused by environmental chemicals and pharmaceuticals to build initial prioritization models of in vivo toxicity. 193 Chemical-response signatures for 87 end points covering molecular pathways pertinent in toxicity were first generated in eight cell systems (primary human cells) for 641 environmental chemicals and 135 pharmaceuticals, followed by computational clustering of the profiling data to determine off-target effects. The measured end points could be closely linked to in vivo outcomes, thereby demonstrating utility of this approach for identifying the potential toxicological liabilities of chemicals in vitro rather than relying on live rodent studies for large numbers of industrial chemicals. The aforementioned ACToR database helps to manage large data sets being generated via the ToxCast program. Xing et al. 194 developed a freely available web server, LTMap, that allows comparisons of global transcriptomic data generated from cells or tissues dosed with candidate drugs against a pregenerated signature database of microarray data sets associated with ~170 compounds. Promising results for DILI prediction have been demonstrated in a few cases, such as improved prediction over animal models using short-term drug dosing in sandwich cultures of PHHs coupled with a high-content imaging classifier 180 and an in silico SAR model that relates chemical structures to the liver side effect data in the LTKB. 195
A few groups are engaged in creating mechanistic computational models of DILI using available data from the literature. For instance, DILIsym software simulates the mechanistic interactions and events from drug administration through the progression of liver injury and regeneration. 196 This software can model some aspects of mitochondrial toxicity, bile acid toxicity, and innate immune responses. Using DILIsym coupled with in vitro data, methapyrilene toxicity was correctly predicted to occur in rats but was not apparent in the simulations for humans and mice, which is consistent with the literature. 197 Strand Life Sciences (Bengaluru, India) has commercialized “Virtual Liver” software that contains a mathematical model of normal liver physiology with pathways for oxidative stress, cholestasis, steatosis (fat accumulation), energy depletion, and cytoskeletal maintenance. 198 When coupled with assays on specific cellular targets, this model provides mechanistic insights into how a drug affects the liver, specifically for the aforementioned pathways implicated in DILI. One key advantage of the Virtual Liver and DILIsym models is that, because they have not been “trained” on specific classes of compounds (as with toxicogenomics), they are not biased/limited to specific classes. Instead, such models are built by modeling gene and protein interaction information curated from the literature (typically manually) and modeled as ordinary differential equations. However, it is not always trivial to integrate data from different literature sources using a host of different species, culture/tissue model systems, and experimental approaches. Despite these limitations, integrating such biological information continually into mechanistic computational models is likely to yield important advances in the field of in silico predictions of DILI.
Common Strategies and Challenges in Designing and Implementing Liver Culture Systems for Drug Screening
Microenvironmental Determinants of Human Hepatic Functions
While animals provide very useful information prior to human clinical trials, there are classes of drugs for which animal liver responses are significantly different from human livers. 6 Thus, the use of human-relevant in vitro models has become necessary in preclinical drug development. Human hepatocytes reside in a complex in vivo microenvironment, containing multiple types of NPCs, complex mixtures of ECM, and dynamic gradients of both soluble and insoluble factors. PHHs were first isolated almost 4 decades ago. Since then, as illustrated with examples above, several attempts have been made to mimic one or more of these cues in vitro depending on the application at hand and throughput desired.11,12,94 As a result of such research, the use of in vitro liver models in drug development and other life science applications has steadily increased, especially over the past 15 years.11,12 While it is not feasible here to summarize important characteristics of every liver model that has been developed to date, we provide key features of a few commercial engineered liver models in Table 1 .
Features of Commercially Available Engineered Liver Models.
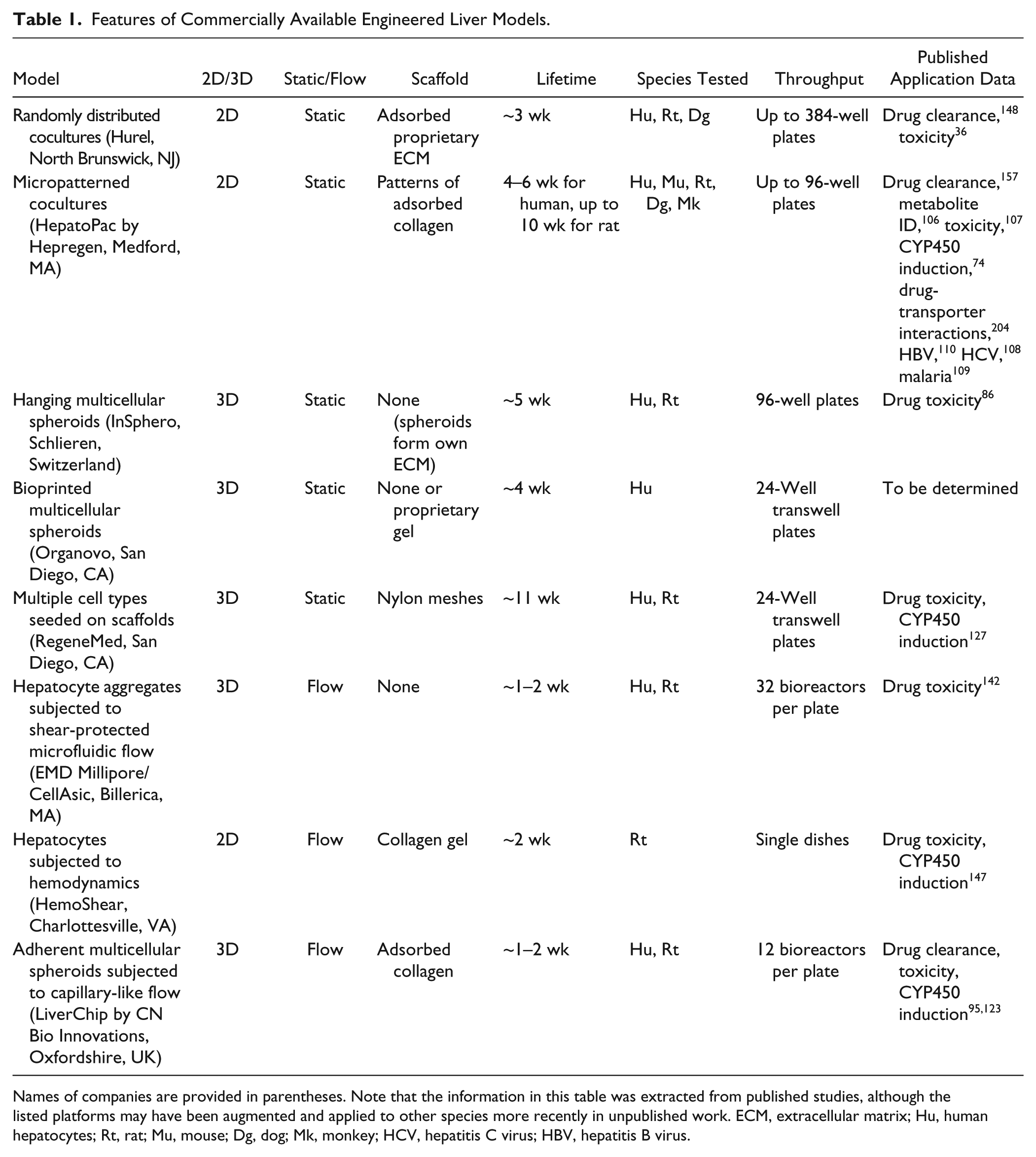
Names of companies are provided in parentheses. Note that the information in this table was extracted from published studies, although the listed platforms may have been augmented and applied to other species more recently in unpublished work. ECM, extracellular matrix; Hu, human hepatocytes; Rt, rat; Mu, mouse; Dg, dog; Mk, monkey; HCV, hepatitis C virus; HBV, hepatitis B virus.
For drug toxicity screening and drug metabolite identification, conventional cultures (i.e., ECM sandwich) have underperformed with ~30% to 50% prediction of clinical outcomes.8,162,180 We now know that stabilizing the PHH phenotype using contact coculture with NPCs, even non–liver derived (i.e., 3T3 murine embryonic fibroblasts), can significantly improve the sensitivity of toxicity and metabolite detection to between 70% and 75%, which is not obtained using rat hepatocytes in the same culture model dosed with the same drug set.106,107 These improvements are partly due to the ability to chronically dose stable PHHs with drugs to mimic clinical drug dosing regimens as opposed to being limited to dosing over 4 to 24 h in conventional formats. Indeed, a majority of current liver models, including those in the commercial space (i.e., RegeneMed, Hurel, Hepregen, InSphero, Organovo), incorporate one or more NPCs alongside hepatocytes.74,86,127,148 Coculture of hepatocytes with NPCs likely maintains hepatic functions due to the concordant impact on the local ECM, soluble microenvironment, and cell-cell interactions.82,102 Furthermore, there is evidence that coculture of different liver cell types in the same device/well allows better stability and functionality of each cell type as opposed to when they are cultured alone.79,95,121 However, while this “coculture effect” appears to be well conserved across species, not all NPCs induce the same level of functions in hepatocytes. 82 Therefore, the selection of an appropriate NPC type is important to enable high levels of liver functions for weeks to months in vitro. Interestingly, in our experience and that of others, liver-derived NPCs do not fully stabilize the hepatic phenotype, at least on their own. 79
We also know that controlling the extent and placement of cell-cell interactions (both homotypic and heterotypic) in vitro either via micropatterning tools or via assembly of spheroids of controlled dimensions plays an important role in optimizing hepatocyte functions and enabling downstream applications that were previously intractable.74,86,96,123 Such has been the case, especially in infectious diseases, where micropatterned cocultures of hepatocytes and embryonic fibroblasts were able to sustain pathogen (HCV, HBV, malaria) infection and replication over randomly distributed mono- or coculture formats.108–110 Culture media formulation has also proven to be critical in inducing higher functions and longevity of hepatocyte culture models.11,81,199 Furthermore, optimal media formulations can vary considerably depending on the types of NPCs used alongside PHHs. Since PHHs are highly metabolically active, their oxygen uptake through the culture medium has been shown to be important in the rate at which they recover in vitro to higher functional levels following the trauma of isolation. 200 Thus, optimizing a given media formulation (including oxygen diffusion through the media height being used) depending on the types of cells being cultured simultaneously is a critical exercise to yield higher functioning and longer lasting cultures. However, in our experience, supplementation of culture medium is necessary but not sufficient to yield stable liver cultures, thereby requiring the use of organized homotypic and heterotypic cell-cell contacts.
Even with the advances made in hepatocyte-NPC cocultures, such devices are still not able to predict ~25% to 30% of drug metabolites and toxins (i.e., sensitivity of 70%–75%).106,107 It remains an open question as to why this is the case. Is it due to the inability to dose cultures with drugs for several months, the lack of NPCs (i.e., macrophages, T cells) to further modulate the hepatic response to drugs, or the use of animal-derived ECM over those from human livers, or is the drug concentration inside the PHH not quite at in vivo levels due to lack of flow and/or presence of other tissue types that affect bioavailability and biodistribution parameters? For detecting metabolites that are missed by cultures, extrahepatic metabolism as enabled by organs-on-a-chip approaches may ultimately be necessary. It is likely that engineering the aforementioned factors will improve the prediction capacity of in vitro liver models, as investigators have evaluated the effect of such cues individually on liver-drug interactions and found them to be beneficial over controls.11,12,95 As advances are made in this space, the necessary but sufficient cues will emerge, and we anticipate consensus will be reached as to the optimal configuration for liver cells, both PHHs and their liver-derived NPC neighbors.
There seems to be a shift in the field from strategies that are solely biomimetic (i.e., culturing hepatocytes in sandwich ECM to mimic the Space of Disse) to culture models that may not necessarily mimic the native hepatic microenvironment (i.e., disorganized spheroids, cocultures with embryonic fibroblasts) but can yield high levels of liver functions and longevity of phenotype for several weeks in vitro. Such models have then been optimized for specific downstream applications (i.e., metabolite profiling, drug toxicity profiling) in different phases of drug development. Whether 3D architecture in the form of spheroids/aggregate-based cultures and cocultures will yield greater advances than engineered 2D models remains an open and untested question, partially because different groups do not have access to each other’s platforms to be able to make side-by-side comparisons. Furthermore, normalization of data (i.e., based on cell number, protein, or RNA levels) is not carried out consistently across different studies, nor are similar end points used to demonstrate liver phenotype so as to enable better comparisons across models by analyzing findings in the literature. Pharmaceutical companies are testing various culture models in their laboratories but do not always publish their results. That being said, highly functional 2D models are being used to build the first generation organs-on-a-chip devices. 151 Thus, it appears that high liver functionality, stability of phenotype for prolonged times in vitro, and higher in vitro–to–in vivo predictive power constitute factors that are of paramount importance when developing tissue models for pharmaceutical practice.
The focus is now on human liver cells as opposed to animal liver cells since the latter have different culture requirements and are ultimately not predictive of human outcomes for many drug classes. PHHs (both freshly isolated and cryopreserved) are now available commercially through several vendors internationally. Hence, it is important that the plethora of liver models being developed be optimized for use with PHHs if they have not been already. Otherwise, whether such models are truly translatable to human relevancy will remain a reasonable source of doubt. We anticipate that, as a nearly inexhaustible source of cells from multiple donors, iHeps may provide a good surrogate for PHHs to build the initial devices in many cases, and they may perform as better alternatives to cancerous cell lines from single donors. However, comparison to freshly isolated PHHs will be necessary to gauge the maturity of the iHeps. 201
Practical Considerations
One important consideration in the design of next-generation liver devices is the type of material used (i.e., scaffold type, tubing for microfluidics) to interface with the cells. There have been instances where a large majority of a dosed drug can bind to the material and not reach the cells, which may be interpreted as lack of cell-mediated drug metabolism in the absence of proper cell-free control devices. 120 We also find that some drugs bind to tissue culture polystyrene, and thus cell-free control wells need to be carried out to determine effects of such binding on rates of drug clearance obtained from PHH cultures. The Griffith group has paid particular attention to this issue by using materials in their perfused liver bioreactor that do not significantly bind lipophilic molecules, including hormones that the cells need to function optimally. 95 In another cell-based approach, Schimek et al. 202 coat their microchannels with primary human dermal microvascular endothelial cells, thereby preventing drug-related material from binding to the plastic. These examples underscore the importance of quantitatively understanding both cell-drug and material-drug interactions in engineered tissue models.
Human liver gene expression and functions relevant for drug screening should be characterized to the extent possible. Global gene expression profiles (i.e., Affymetrix [Santa Clara, CA] microarrays) in the engineered liver models are useful to analyze diverse pathways expressed over time in culture. Reverse transcription–quantitative PCR (RT-qPCR) (i.e., TaqMan assays from Life Technologies) can also be used to confirm findings from the microarrays and for more routine gene expression analyses at multiple time points. However, multiple functional analyses (i.e., activities of drug metabolism enzymes) are important for pharmaceutical adoption of new liver models. We have provided in Table 2 a listing of hepatic end points/assays that, in our experience, constitute a good initial validation of a liver model in conjunction with the aforementioned global gene expression analysis. For those liver models that include liver NPCs, Table 3 lists some common ways to appraise the phenotype of these cells. It is important that both gene expression and functional levels in engineered liver tissues be compared with those obtained from intact liver tissue when available and freshly isolated liver cells. Finally, on the basis of our own work with several major pharmaceutical companies, we present here an initial validation strategy for engineered liver model systems in key applications during drug development ( Table 4 ).
Human Hepatocyte Functions That Can Be Measured Over Time in Engineered Liver Models.

Vendors that provide assay kits and/or reagents and associated protocols are listed in parentheses where available. LC-MS (liquid chromatography–mass spectrometry)–based assays can be carried out by analytical vendors such as Integrated Analytical Solutions (Berkeley, CA) and Cyprotex (Watertown, MA).
Assessment of Phenotype of Human Liver-Derived Nonparenchymal Cell (NPC) Types Over Time in Engineered Liver Models.
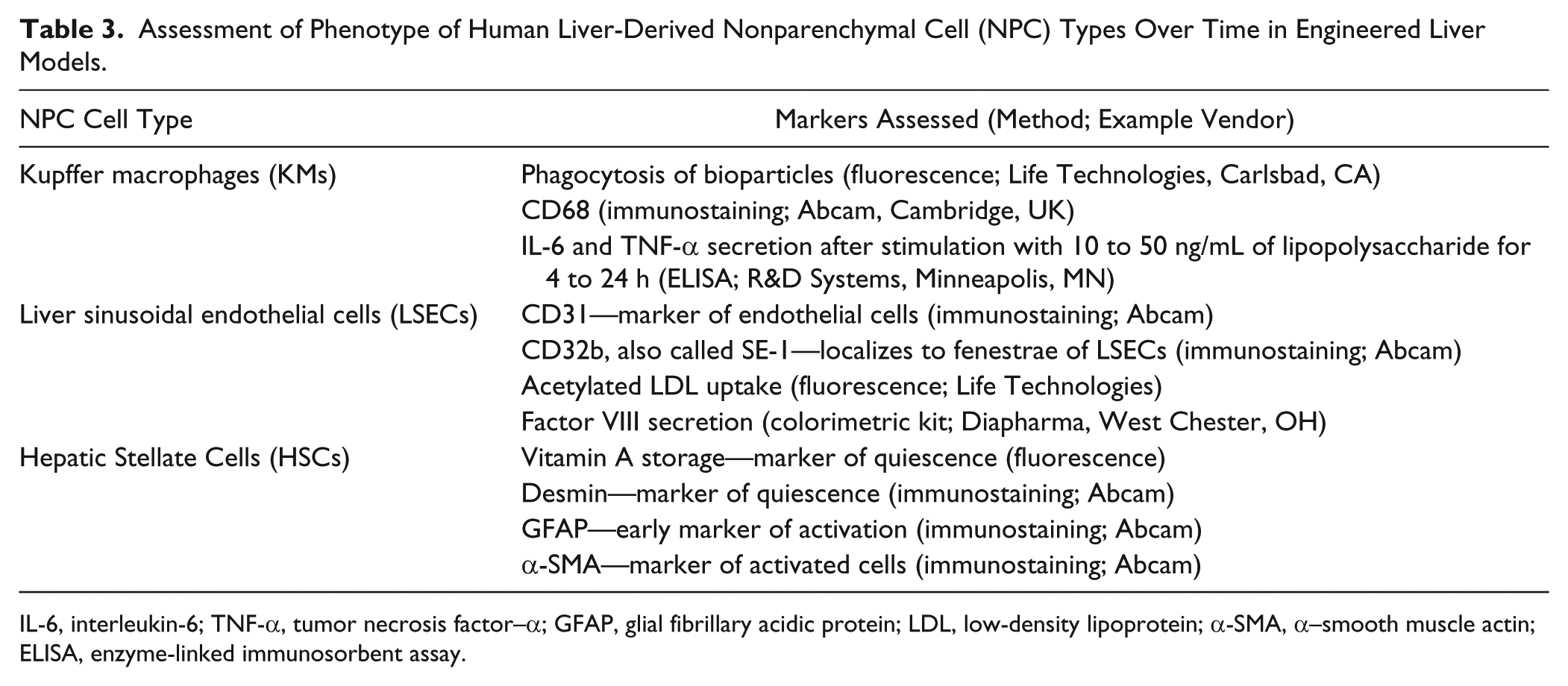
IL-6, interleukin-6; TNF-α, tumor necrosis factor–α; GFAP, glial fibrillary acidic protein; LDL, low-density lipoprotein; α-SMA, α–smooth muscle actin; ELISA, enzyme-linked immunosorbent assay.
Proposed Initial Validation of Engineered Liver Models for Key Applications during Drug Development.

ALT, alanine aminotransferase; ATP, adenosine triphosphate; GSH, glutathione; LC-MS/MS, liquid chromatography/tandem mass spectrometry; LDH, lactate dehydrogenase.
Cost is a critical parameter of drug development, where companies are often competing vigorously to bring a new therapeutic to the market. Many arguments have been made that pharmaceutical companies will save millions, if not billions, of dollars by using in vitro models to eliminate toxic compounds from the pipeline and/or to design better drugs prior to reaching human clinical trials or the marketplace where failures can be very costly.1,2 However, in our experience, while pharmaceutical companies are embracing such a paradigm as evident in the increased use of in vitro models over the past decade, ultimately budgets are controlled by different members within the organization in different parts of the pipeline, and thus, it is not always trivial to move money around to accommodate one part of the pipeline over another. Therefore, it is critical that the cost-benefit ratio of a given liver model be clearly articulated to pharmaceutical managers with the appropriate validation data. The validation data acquired by a vendor (preferably presented as a peer-reviewed set of publications) may be promising but not sufficient for a pharmaceutical audience working on therapeutics for specific diseases. Thus, the first few projects with a pharmaceutical client are often geared toward collection of validation data of that particular client’s compounds. Such customized validation for each major client can definitely slow down the adoption of a new culture model by the overall pharmaceutical industry; however, with many competing liver models coupled with the proprietary drug sets of specific companies, such validation exercises appear to be necessary based on our experience. That being said, organizing a consortia of pharmaceutical companies and academic institutions that can agree on sets of drugs as well as end points for validating liver culture models for specific applications is likely a worthwhile endeavor to improve the efficiency of platform adoption for screening live compounds prospectively. There are some examples of such consortia for drug/chemical screening,6,188 but it has not been implemented consistently and on an ongoing basis.
The throughput of a platform is also essential to be able to test more than just a few compounds, especially because timelines during drug development are quite tight. It is for this reason that multiwell plates (i.e., 384-well format), which can be coupled with robotic fluid handlers, are preferred for drug screening. Furthermore, the cost for testing each drug is lowered if more drugs can be tested on a given plate of cells, allowing more drugs to be put through the cell-based screening process. However, as is often the case with higher throughput and lower cost, one may lose some physiological relevance, as is the case with cancerous cell lines seeded in multiwell plates. Thus, compromises have to be made in a given part of the drug development pipeline, and suitable liver models for the specific question being asked may not suffice for other questions. The relay method described earlier with suspension pooled hepatocytes to predict clearance of low-turnover compounds is one of many examples where nonengineered liver models, in some cases with cancerous cell lines,33,36,39,203 continue to be used to cut down cost during drug development and improve throughput, while proving effective for a given purpose within the investments already made in cells and technology infrastructure in pharmaceutical companies.
Future Outlook
In our view and that of others,11,120 a single engineered liver model will likely not serve as the panacea for the entire drug development pipeline. In fact, one could make the argument that reliance on a single in vitro model for all the compounds is inefficient and not cost-effective for the various stages of a drug development pipeline. In vitro models, by design, afford the investigator an opportunity to develop an assay with sufficient complexity that is fit for the purpose at hand but not with so much complexity that results are difficult to interpret and/or the necessary throughput is not attained. Thus, as described above, we anticipate that different liver models will continue to serve the specific needs of the pharmaceutical industry depending on the complexity of questions being asked, the need for varying throughput, and budgetary constraints.
As an example of how different culture models of varying complexity could serve in drug development, one can look at the critical task of screening for adverse effects/toxicity of new drug candidates. For early drug discovery, hepatic cell lines and/or iHeps may suffice to identify the highly toxic compounds that can either be discarded or modified structurally to reduce the toxicity. Later in drug development, more stable cultures of PHHs and liver NPCs (i.e., micropatterned), still in a 2D multiwell format for higher throughput screening, could be used to determine if chronic drug dosing may affect one or more liver cell types relative to control compounds. If a drug makes it through these two stages without toxicological liabilities being identified, it could then be tested in a lower throughput but higher content (i.e., via imaging, transcripotomics, metabolomics, proteomics) 3D human liver model that has as many of the liver microenvironmental cues as can be incorporated into a system while maintaining its reproducibility and batch-to-batch consistency. Such a model could also be interfaced with other tissue models to determine how the liver might modulate toxic effects of drugs on other organs and vice versa. Finally, select compounds could then be tested in animals as currently deemed necessary by the FDA to obtain a measure of how the drug affects a living organism, with the caveat that if differences are seen between a drug’s effect on animals as opposed to the human liver models, those could very well be due to species-specific differences and not just due to in vitro artifacts (i.e., false positives). Thus, comparison of drug metabolism and toxicity in multiple species in vitro may be needed upfront to select the appropriate animal model for evaluating the toxicity potential for a given class of compounds in vivo. For that reason, development of in vitro animal liver models remains an important endeavor for drug testing. However, there will likely continue to be cases where engineered human liver models are the only means by which a drug’s toxic effects can be studied in preclinical testing. We personally have come across and aided in such cases, especially after a drug had gone all the way into clinical trials and caused liver enzyme elevations in patients but not in preclinical animal species.
For drug metabolism studies, it does not seem likely that the next-generation liver models will replace conventional models (i.e., cell-free microsomes, suspension hepatocytes, and plated hepatocyte cultures) entirely but provide complementary tools to investigate phenomena where conventional models may not suffice. For instance, conventional models may provide initial information on route of metabolism, prediction of clearance, major metabolites generated, and enzyme induction. Then, lead candidates could be tested on engineered liver models to determine effects of chronic drug dosing on such outcomes, as has already been shown to be useful for clearance prediction and metabolite generation.106,157 Even enzyme induction and prediction of DDI potential could benefit from chronic dosing of liver models as we have described earlier. Testing the remaining compounds further on organs-on-a-chip could allow assessment of how other tissues besides the liver affect drug disposition, including interaction with transporters present ubiquitously in many organ systems. Finally, very few studies could be carried out in animal models to confirm findings generated by the in vitro models, but while taking into account any species differences that could confound interpretation. However, when integrated assessment of drug metabolism and toxicity need to be carried out to determine how one affects the other, especially when the drug is closer to the clinic, and/or its effects in the clinic need to be investigated mechanistically, engineered liver models that provide higher sensitivity would likely be the only option, especially if animal models previously failed to predict human-relevant drug effects.
In conclusion, the progress that has been made and continues to be made at an enhanced pace in building next-generation liver models will provide pharmaceutical companies with more predictive choices, which will likely reduce cost of drug development significantly, reduce usage of animals (and potentially eliminate it in the future), and reduce harm to human patients in clinical trials and in the marketplace. These models will also be useful to discover novel molecular targets for diseases that affect the liver, allowing development of more efficacious therapeutics with fewer side effects. Such is the promise of engineered liver culture models, and we are already realizing at least part of that promise with several of the aforementioned examples in the practice of drug development.
Footnotes
Acknowledgements
We acknowledge Dr. Sangeeta Bhatia for helpful discussions.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Salman R. Khetani is stock holder in Hepregen Corporation, which has exclusively licensed the Micropatterned Coculture (MPCC) Technology from M.I.T. for drug development applications.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding was provided by Colorado State University and NSF CAREER grant (CBET-1351909) to S.R.K.