
Abstract
Transcription activator–like effectors (TALEs) are becoming powerful DNA-targeting tools in a variety of mammalian cells and model organisms. However, generating a stable cell line with specific gene mutations in a simple and rapid manner remains a challenging task. Here, we report a new method to efficiently produce monoclonal cells using integrated TALE nuclease technology and a series of high-throughput cell cloning approaches. Following this method, we obtained three mTOR mutant 293T cell lines within 2 months, which included one homozygous mutant line.
Introduction
Transcription activator–like effectors (TALEs), discovered in the plant pathogen Xanthomonas sp., have attracted much attention because of their high specificity for DNA binding and varied functionality for genome engineering.1,2 The nucleotide recognition region of TALEs is known as the repeat variable diresidue (RVD), which determines the nucleotide-binding specificity (NI=A, HD=C, NG=T, NN=G or A). 3 TALE nucleases (TALENs) are constructed by fusing the TALE DNA-binding domains with the nuclease FokI, 4 and the current synthesis methods have already been commercialized and improved.3,5,6
To date, TALENs have successfully been applied to edit target genes in various species, such as Drosophila, zebrafish, mouse, and even human cells.7–10 To efficiently generate genome editing products for human cells, DNA double-strand breaks (DSBs) have to be introduced at specific chromosome sites by TALENs. 10 Given that the targeting efficiency is variable, achieving a gene mutant stable cell line in a simple and effective manner becomes a central problem.
We recently developed a strategy, which integrates TALEN technology and a high-throughput cell sorting approach, to produce a stable cell line with a specific gene mutation ( Fig. 1A ). To illustrate this strategy, we chose mTOR as an editing target because of its critical role in regulating cell autophagy. 11 First, we exploited a magnetic beads–based TALE assembly method to synthesize a group of TALEN vectors to target mTOR 3 and introduced them into 293T cells by transfection. Second, we confirmed the gene editing activity of the TALENs using exogenous or endogenous detection assays. Third, we performed cell cloning screening using high-throughput instruments. Finally, we identified monoclonal cells and achieved three stable cell lines containing the mutant genes.
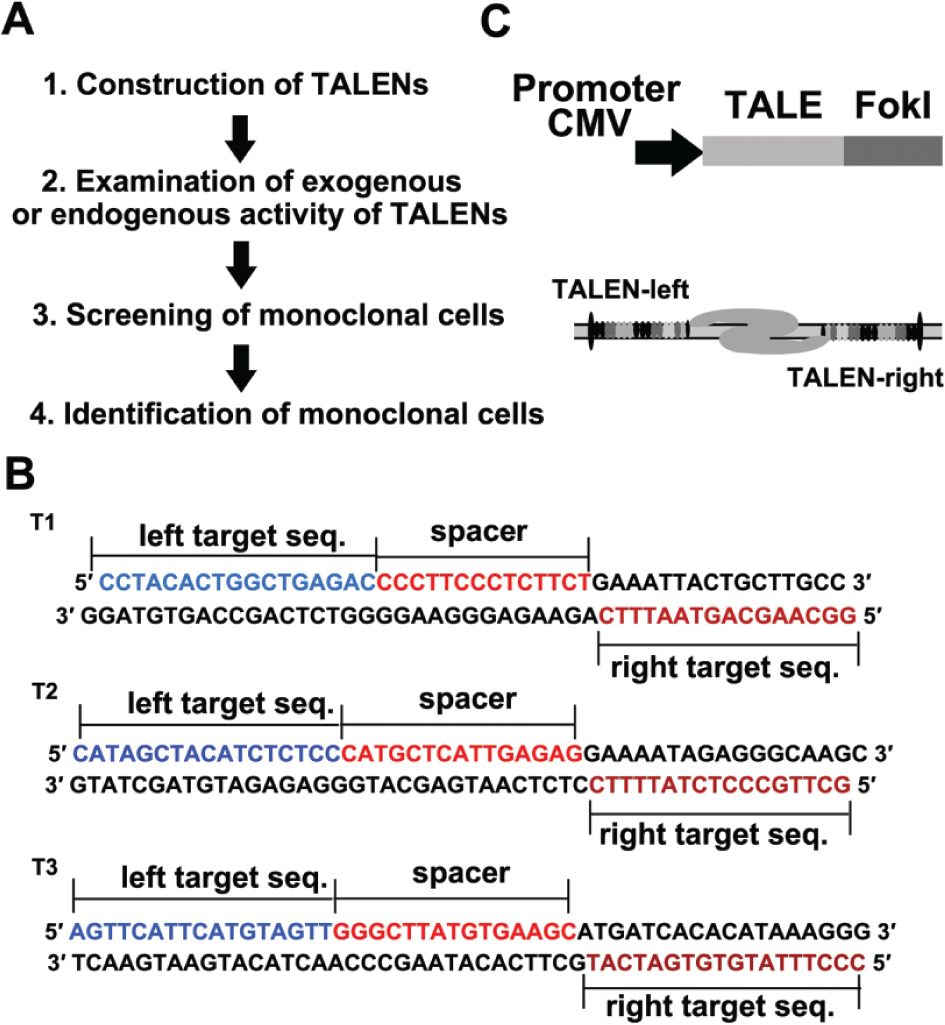
Construction of transcription activator–like effector nuclease (TALEN) vectors. (
Materials and Methods
Cell Culture and Transfection
The 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 0.1 mg/mL streptomycin under humidified conditions in 95% air and 5% CO2 at 37 °C.
The transfection experiments using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) transfection reagent were performed according to the manufacturer’s procedure. The optimized transfection protocol was described as follows: 5 µL transfection reagent/1 µg TALEN-L plasmid/1 µg TALEN-R plasmid in a 35-mm dish.
Design and Construction of TALEN-Expressing Vectors
We used the TAL Effector Nucleotide Targeter 2.0 online software (Iowa State University, USA) to design TALENs targeting the mTOR gene ( Fig. 1B ). 12 Then, we implemented a solid-phase synthesis strategy, 3 which allows a quick and simple purification of the ligation product to construct TALEN vectors. We performed the TALE production by use of a solid-phase gene synthesis approach we reported previously. In brief, we took use of a chip that contains a microwell array and magnetic microbeads coated with streptavidin. Through three steps—monomer ligation, enzymatic digestion, and purification—we can synthesize a group of TALENs in an efficient manner. The proteins are expressed under the control of cytomegalovirus (CMV) promoter ( Fig. 1C ), and they specifically recognize a DNA sequence through RVDs ( Table 1 ).
Transcription Activator–Like Effector Target Site Sequences and the Corresponding RVDs (NI=A, HD=C, NG=T, NN=G).

RVD, repeat variable diresidue; TALEN, transcription activator–like effector nuclease.
Results
Examination of Exogenous or Endogenous Activity
First, we used the luciferase single-strand annealing (SSA) reporter system to easily and rapidly evaluate the editing activity after the TALEN-mediated DSBs. 3 If the TALENs worked well, the luciferase activity (firefly/Renilla) of target gene could be recovered through SSA repair, compared with an empty vector in the negative control experiment (NC). The cells were transfected with the plasmids in the following ratio: TALEN-L plasmid/TALEN-R plasmid/luciferase reporter/Renilla = 1:1:1:0.05, where Renilla was used as the internal control. Twenty-four hours after transfection, cells were lysed in lysis buffer (Promega, Madison, WI) and detected by an automatic microplate reader (Multimode Reader, EnSpire, PerkinElmer, USA) with a dual-luciferase reporter kit (Promega). The luciferase activity was increased by 5-fold upon cotransfection with one of the TALENs ( Fig. 2A ), which indicated that the TALENs have high editing activity on the target sequence of the mTOR genome.
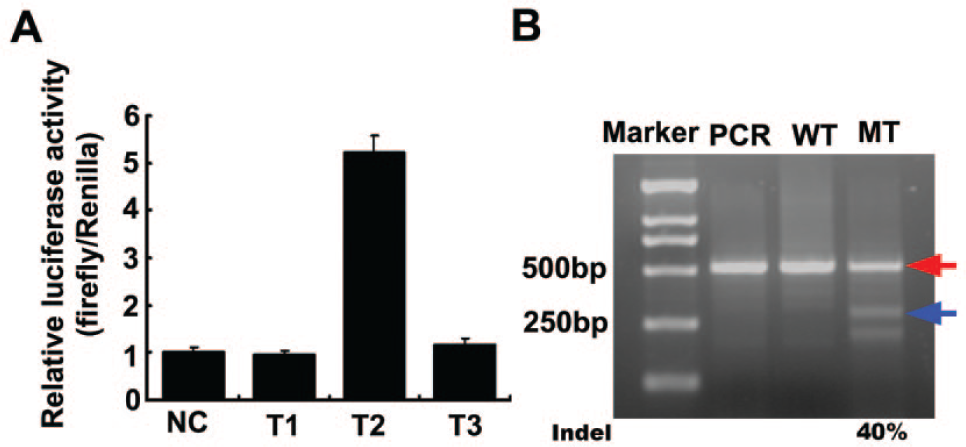
Detection results for editing activity of mTOR transcription activator–like effector nucleases (TALENs). (
Next, we used a T7 endonuclease I (T7EI) assay to intuitively assess the endogenous activity of the TALENs. 6 The cells were transfected in the following ratio: TALEN-L plasmid/TALEN-R plasmid = 1:1. Then the cells were divided into two groups: a mutant group (transfected with TALENs) and a wild-type group (transfected with green fluorescent protein [GFP]). Two days after transfection, cells were lysed in lysis buffer (Takara, Japan) and the reactions were incubated at 65 °C for 10 min then at 95 °C for 15 min. We used 1 µL of the lysate in a 25-µL PCR reaction using MightyAmp DNA Polymerase (Takara). The sequences of primers used for genomic PCR were AGCACCGTCTTTGCAAGTCA (forward) and TCCAGA GCCCACTTGCTTAAC (reverse). The annealing temperature was 58 °C, the elongation time was 30 s, and the cycle number was 30. The PCR products were annealed with themselves, and then 0.5 µL T7EI (View-solid, China) was added and the reactions were incubated at 37 °C for 30 min. The samples were separated on a 2% agarose gel at 100 V. We observed distinct cleaved DNA products in the mutant group but not in the wild-type group ( Fig. 2B ), indicating that the mTOR TALENs have very high endogenous activity. The mutant efficiency reaches about 40% through the following calculation formula: Intensity of cut brands/(intensity of cut brands + intensity of uncut brand)*100%.
Screening of Monoclonal Cells
Once the endogenous editing activity of the mTOR TALENs in 293T cells was confirmed, we used high-throughput instruments to clone 293T cells from the transfected cell pool by a limiting dilution method, 13 and the cell clones were screened by an imaging method.
The screening procedure is described in Figure 3A . First, we trypsinized cells in medium and measured the cell density through an automatic cell counter (Scepter 2.0 Handheld Automated Cell Counter; Millipore, Billerica, MA). Second, we diluted it in a serial cell density—1 × 105, 1 × 104, and 1 × 103 cells per milliliter—and the final density was one cell per 100 µL in 15 mL medium. Third, we dispensed 120 µL medium into one 96-well cell culture plate (Costar, LifeScience, USA) through an automatic cell dispenser (Multidrop Combi; Thermo, Waltham, MA). After approximately 12 days, we screened the wells and captured images of cell clones using a high-content imaging system (ImageXpress; Molecular Devices, Sunnyvale, CA). Finally, we determined the clone number in each well according to the images ( Fig. 3B ). In total, we picked 60 wells with monoclonal cells marked with Arabic numerals 1 to 60, and then these cells were trypsinized. Half of the cell monoclones were seeded in 48-well plates (Costar), and the other half of clones were stored in an EP tube at −80 °C.
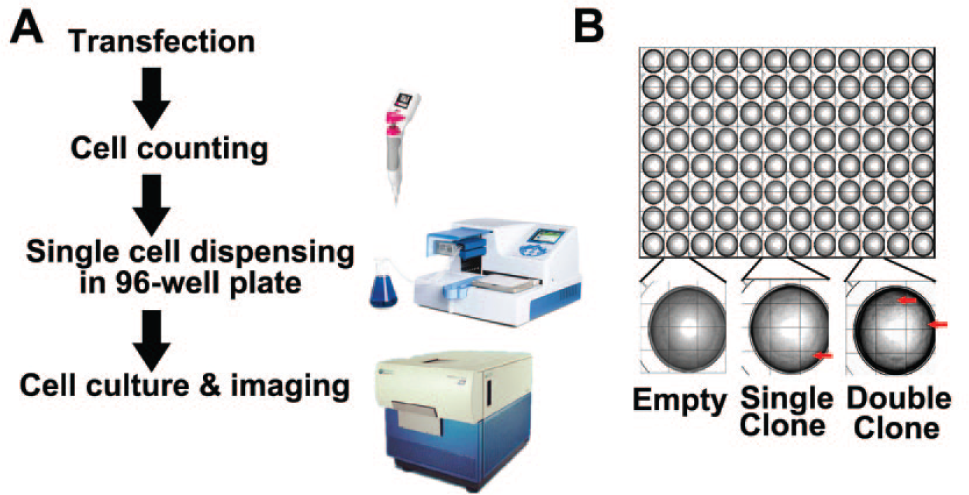
Automatic process of cell cloning screening. (
Identification of Monoclonal Cells
We selected 15 cell clones to examine. Cells from different clones were lysed and assayed with PCR as described in Materials and Methods. Then, each monoclone PCR product was annealed with wild-type PCR product and was followed with T7EI assay as a primary screening. The samples were separated by 2% agarose gel electrophoresis at 100 V. Three clones (No. 3, No. 4, and No. 14) showed mutant sequence ( Fig. 4A ). To confirm the genome sequences of the three cell clones, we implemented a TA cloning sequencing method using the pMD19-T kit (Takara). The sequencing results demonstrated that cell clone No. 3 or No. 4 had a heterozygous mutation, whereas cell clone No. 14 had a homozygous mutation ( Fig. 4B ). To quantify the mutation efficiency in the mTOR genome, we also designed real-time PCR analysis with the following primers: ATGCTCATTGAGAGGAAAATAGAG (FP) and ACCTGCCTTCCACTGTTACTTCT (RP) ( Fig. 4C ). As shown in the real-time PCR results, the mutation efficiency reached around 35% in TALEN-transfected cells. Furthermore, the mutation efficiency reached 50% in the No. 3 cell clone and 100% in the No. 14 cell clone, thus indicating that the former is a heterozygous mutant and the latter is homozygous one ( Fig. 4C ). Thus, we achieved the construction of three mTOR mutant 293T cell lines within 2 months. This method can be applied to other cell lines with other mutant genes. Advanced DNA delivery approaches can be used to improve transfection efficiency, such as electrotransfection, lentivirus, adenovirus, and drug resistance (usually implemented 24 h after transfection).
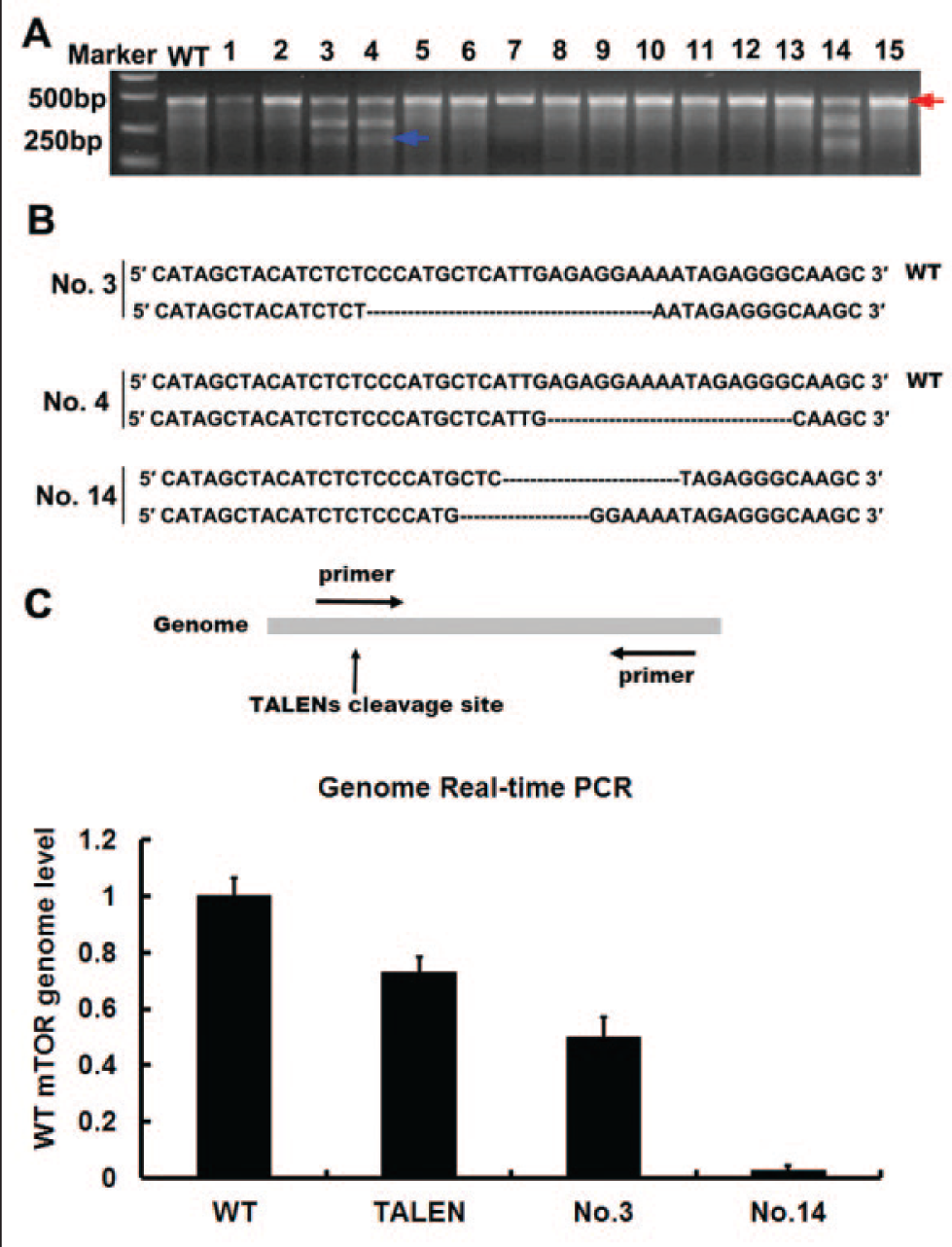
Identification of cell monoclone gene types. (
Discussion
In our TALEN vector construction process, we performed codon optimization in RVDs for Homo sapiens ( Table 1 ). In the transfection protocol, we optimized the proportion of transfection reagent and TALEN plasmids. The optimized transfection protocol was described in Materials and Methods. Through these optimized implementations, endogenous mutant efficiency induced by TALENs reached as high as 40% in the T7E1 assay ( Fig. 2B ) and around 35% in the real-time PCR analysis ( Fig. 4C ).
In our cell cloning screening strategy, we applied several high-throughput instruments, including a cell counter, a cell dispenser, and a high-content imaging system. The implementation of these commercially available instruments can dramatically enhance the stability and accuracy of the screening process. Compared with a traditional hemocytometer, the cell counter not only counts cell number rapidly and accurately but also excludes the very small sized cells, which are generally apoptotic cells or cell debris. Thus, the application of a cell counter can increase the survival ratio of monoclonal cells. In addition, scientists have preferred cloning cylinders with the cell cloning method, 14 which is a complicated and time-consuming approach. Recently, fluorescence-activated cell sorting (FACS) also has been used in sorting, but we found that the cells sorted from FACS required more time for cloning, because of medium containing sheath flow liquid. In our strategy, we used a limiting dilution method. The cell dispenser allowed the dispensing of the medium containing cells in a very short time, so the cells were appropriately distributed in the 96 wells. Besides these instruments, we also used a high-content imaging system, which can facilitate cell clone recognition in an easy and accurate manner. In total, we generated 60 cell clones out of 96 wells within 12 days at a low cost, with the requirement of no additional instruments, such as cloning cylinders or FACS. Thus, our strategy showed superiority to the traditional methods in monoclone screening, such as being less time-consuming, having a lower cost, and having more monoclone formation ( Table 2 ).
Comparison of the Traditional Approaches with Our Strategy in Monoclonal Cell Screening.

Last, we identified three mutant cell clones out of 15 monoclonal cells, including two heterozygous mutant clones (No. 3 and No. 4 cell clones) and one homozygous mutant clone (No. 14 cell clone). These results indicate that this method can be used to produce gene mutant cell lines in a robust and efficient manner.
Perspective
A few of the high-throughput assembly approaches of custom TALENs constructs have also been developed.3,5 In this work, we exploited the solid-phase gene synthesis of TALENs and presented an efficient method of producing a stable cell line with specific gene mutants. More recently, type II clustered regularly interspaced short palindromic repeats (CRISPRs) have been demonstrated as an alternative approach on genome editing in a variety of mammalian cells and model organisms.15–18 We believe that this method can be easily adapted to CRISPRs and dramatically influence the fields of biomass production, stem cell, cell therapy, or engineering.
Footnotes
Acknowledgements
We thank Dr. Fengfeng Zhuang at Beijing View-Solid Biotechnology for insightful discussions and generous gifts for plasmids.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by projects of MOST (Grant No. 2011CB809106, 2013CB917803, 12AA020103) and NSFC (Grant No. 81030040, 31371443), as well as the Center for Age-related Diseases (CAD), Peking University.