
Abstract
Aptamers are oligonucleotides that can bind to various nonnucleic acid molecular targets in a high affinity and specificity. As an emerging class of therapeutic agents, aptamers offer an unparalleled advantage over other classes of therapeutic agents: the possibility to rationally regulate the therapeutic activity of aptamers. Most existing strategies for regulating the aptamer activity have a limited specificity and/or reversibility. Herein we report a simple, generic strategy to simultaneously achieve specificity and reversibility by exploiting the spontaneous conformational change of hairpin oligonucleotides upon the specific recognition of nucleic acid effectors. The effector-responsive hairpin oligonucleotide consists of a sensing loop that recognizes a particular nucleic acid effector, an aptamer stem that inhibits a certain therapeutic target, and an antidote stem that is complementary to the aptamer. Upon the introduction/removal of the effector, the hairpin oligonucleotide undergoes a conformational change that activates/deactivates the aptamer’s inhibiting activity on the therapeutic target. This new strategy has been demonstrated with an anticoagulant aptamer that binds and inhibits human α-thrombin.
Introduction
Aptamers are oligonucleotides that can bind tightly to specific molecular targets. With the development of the SELEX (Systematic Evolution of Ligands by Exponential Enrichment) process,1–3 aptamers have been generated against a great variety of therapeutic targets.4,5 Aptamers bind to their targets in a specificity and affinity comparable to that of antibodies.4–6 As an emerging class of therapeutic agents, aptamers offer certain advantages over antibodies due to their nucleic acid nature. First, aptamers are produced from chemical synthesis; therefore, they are low cost and highly reproducible. Second, their immunogenicity is low or none, and their stability and bioavailability can be improved with chemical modifications.7,8 Third, but most important, the therapeutic activity of aptamers can be facilely inhibited by antisense oligonucleotides (i.e., antidotes). Therefore, it is possible to regulate the activity of aptamers in a rational manner for safer and more controllable therapeutics.
Recently, several types of strategies to rationally regulate the activity of aptamers have been developed. In the first type of strategies, the aptamer-specific antidotes are introduced when and where the activity of aptamers needs to be reversed. The efficaciousness of this strategy has been demonstrated in both the test tube and several animal models.9–11 In the second type of strategies, light-triggered activation 12 /deactivation 13 is accomplished with caged aptamers/antidotes. The activity of caged aptamers/antidotes is temporarily masked by photolabile groups and can be restored upon irradiation. The light-controlled regulation is usually irreversible, but a reversible light-controlled regulation was also reported recently. 14 In the third type of strategies, a molecular automaton is used to control the release of aptamers or antidotes upon the presence of particular nucleic acid effector molecules. These strategies are usually implemented with restriction enzyme15,16 or DNA/RNAzyme.17,18 For the first and second types of regulation strategies, their specificity relies on the specific delivery of antidotes or light. For the third type of regulation strategies, a high specificity can be achieved at the molecular level; however, the involvement of nucleic acid–cleaving enzymes limits its reversibility.
Herein we present a simple strategy for achieving the reversibility as well as the specificity in the regulation of aptamer activity. This strategy uses the spontaneous conformational change of hairpin oligonucleotides upon the specific recognition of nucleic acid effectors. The effector- responsive hairpin oligonucleotide consists of a sensing loop that targets a specific nucleic acid effector, an aptamer stem that can inhibit a therapeutic target, and an antidote stem that can anneal and inhibit the aptamer. When the effector is introduced or removed, the hairpin oligonucleotide undergoes a conformational change, thus activating/deactivating the therapeutic activity of the aptamer. To prove the concept of this new strategy, it was applied to a well-studied anticoagulant aptamer that binds and inhibits human α-thrombin. The feasibility, specificity, and reversibility have been demonstrated with a gel shift assay and a fibrinogen clotting assay.
Materials and Method
Materials
All the oligonucleotides used in the experiment were synthesized by Integrated DNA Technologies Inc. (Coralville, IA). Upon being received, the oligonucleotides were resuspended in nuclease-free water and stored in aliquots at −20 °C until use. The concentration of the oligonucleotides was determined by measuring the absorbance at the wavelength of 260 nm on a UV/Visible Spectrophotometer (Hewlett-Packard, Palo Alto, CA). The precast polyacrylamide gel and SYBR Gold were obtained from Invitrogen (Carlsbad, CA). Human thrombin and fibrinogen were purchased from Sigma-Aldrich (St. Louis, MO). If not specified, all other reagents were also purchased from Sigma-Aldrich. In most experiments, 1× binding buffer (140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 20 mM Tris [pH 7.4]), whose composition is similar to the buffer used in the selection of the original 15-nt antithrombin DNA aptamer, was used.
Gel Shift Assay
The hairpin oligonucleotides were mixed with the nucleic acid effectors in desired molar ratios. The mixtures were heated at 90 °C for 5 min and cooled to 25 °C at 0.2 °C per 20 s with a PCR Minicyler (MJ Research, Waltham, MA) and then stored at 4 °C until use. The preannealed hairpin-and-effector solutions were incubated with thrombin in 1× binding buffer at room temperature for 1 h before loading to the gel. The electrophoresis was performed on a precast 20% natural polyacrylamide gel in 1× TBE buffer supplemented with 5 mM KCl and 1 mM MgCl2. The gel was prerun for 15 min at 9.7 V/cm before the sample loading and then run at the same voltage for 15 min, followed by a 1-h run at 7.3 V/cm. The gels were stained with 1:10 000 SYBR Gold for 45 min and then imaged by a Molecular Imager FX Pro Plus laser scanner (Bio-Rad Laboratories, Hercules, CA) with excitation at 488 nm and emission at 530 nm. The gel images were processed and quantified with ImageJ (National Institutes of Health, Bethesda, MD).
Fibrinogen Clotting Assay
As a coagulation protein, thrombin cleaves soluble fibrinogen into an insoluble fibrin clot. In this assay, the change in the solution opacity associated with the fibrinogen cleavage was used to quantify the activity of thrombin. Fibrinogen was dissolved in 1× binding buffer by gently inverting and shaking instead of vortexing, thus preventing the breakdown of the fibrinogen molecule. The mixtures of hairpin oligonucleotide, effector, and thrombin were prepared as described in the gel shift assay. These mixtures were first mixed with fibrinogen by pipetting, and the resulting solutions were then quickly added into a half-area 96-well plate (100 µL per well). Careful attention should be paid to avoid introducing bubbles in the well. Absorbance at 400 nm was recorded every 2 min for 2 h on a microplate reader (Molecular Devices, Sunnyvale, CA). A linear fitting was performed on the raw data to obtain the change rate of absorbance, which was then used as a measure of the fibrinogen-cleaving activity of thrombin.
Results
Working Principle of Effector-Responsive Hairpin Oligonucleotides
As illustrated in Figure 1 , an effector-responsive hairpin oligonucleotide is constructed with three components: the sensing loop, the complementary aptamer, and antidote stems. The sensing loop recognizes a specific nucleic acid effector, and the aptamer is against a certain therapeutic target. In the absence of the effector, the hairpin oligonucleotide will remain in the “inactive” state, which allows the antidote to bind to the aptamer, thus preventing the interaction of the aptamer with the therapeutic target. However, in the presence of the effector, hybridization of the effector to the sensing loop will force the hairpin oligonucleotide to transit from the “inactive” to the “activated” state, which separates the antidote from the aptamer, consequently activating the aptamer’s activity on the therapeutic target. By taking advantage of the spontaneous conformational change of hairpin oligonucleotides upon the specific molecular recognition of nucleic acid effectors, the current strategy provides a simple way to realize the specific and reversible modulation on the aptamer activity.
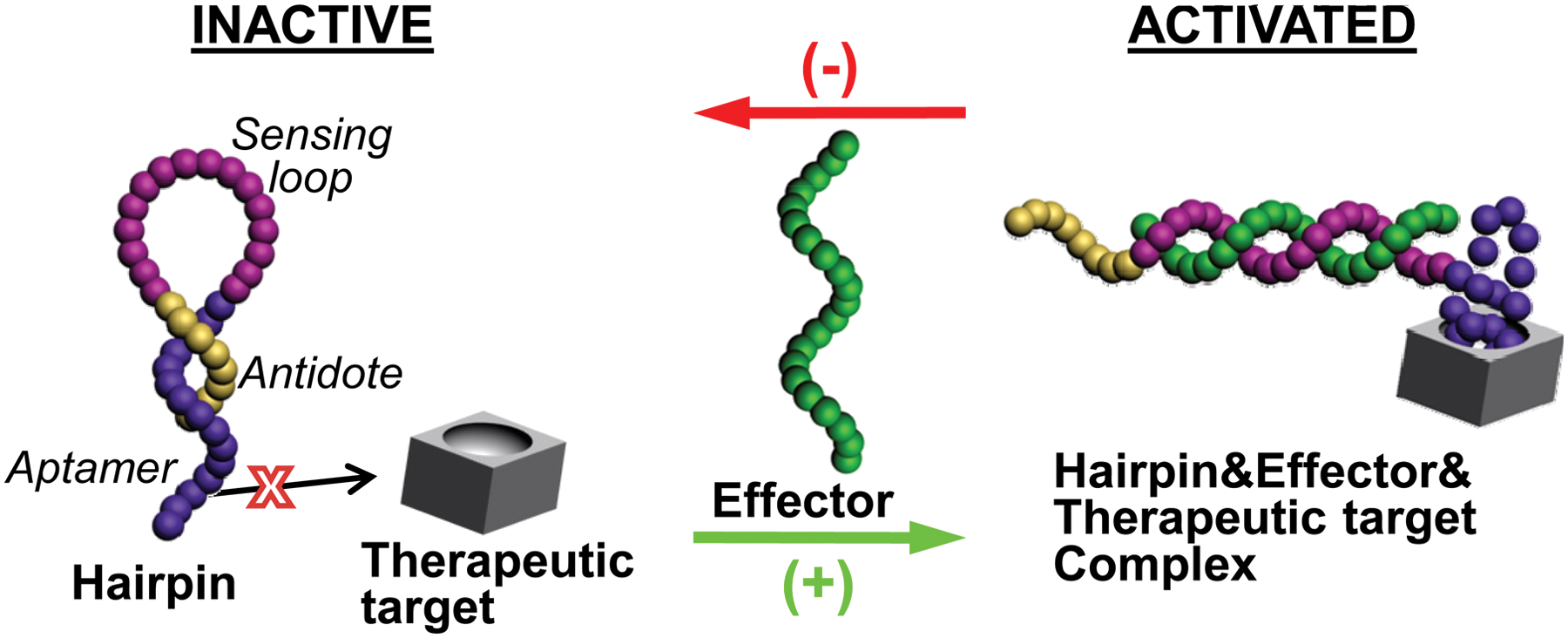
Schematic representation of the working principle. The effector-responsive hairpin oligonucleotide consists of a sensing loop that is complementary to a nucleic acid effector, an aptamer stem that can inhibit a therapeutic target, and an antidote stem that can anneal to the aptamer. Upon the introduction/removal of the effector, the hairpin oligonucleotide undergoes a conformational change that activates/deactivates the aptamer.
Design of the Hairpin Oligonucleotides
To demonstrate the feasibility, this strategy was applied to a well-studied anticoagulant aptamer 19 that binds and inhibits human α-thrombin. Thrombin is a key serine protease in the coagulation cascade 20 and a potent agonist for platelet activation. 21 This 15-nt single-stranded antithrombin DNA aptamer forms a 3D intramolecular structure consisting of three loops and two G-quartets, which were shown to be critical for the activity of this aptamer.22–24 Therefore, an antidote that is complementary to the G-quartet-forming region of the aptamer was chosen here to minimize the remaining aptamer activity in the “inactive” state. To facilitate the future intracellular study, instead of a nucleic acid biomarker indicating a certain disease status, the mRNA of green fluorescent protein (GFP) was used as the effector. The initial design of the hairpin oligonucleotide HP1 consists of a 20-nt sensing loop that targets the GFP mRNA and two complementary stems: the 15-nt antithrombin aptamer and an 8-nt antidote ( Table 1 ). Because modification of the original aptamer sequence tends to cause a loss in the aptamer affinity and specificity,25–27 another hairpin oligonucleotide, HP2, was designed to decrease this effect by inserting a stretch of irrelevant nucleotides “AAAAA” as a spacer between the sensing loop and the aptamer stem.
Sequences of the DNA Strands Used

For hairpin oligonucleotides HP1 and HP2, the underlined italics is the antidote stem, the bold is the sensing loop, the underlined is the aptamer stem, and the normal font is the spacer.
For effectors E1 and E2, the region targeted by the sensing loop of HP1 and HP2 is in bold italics.
E1-C and E2-C are the complementary sequences of E1 and E2, respectively.
TBA is the sequence of the original 15-nt thrombin-binding aptamer.
Binding Tests
Gel shift analysis was first performed to investigate how the nucleic acid effector would affect the binding affinity between the hairpin oligonucleotide and the therapeutic target thrombin. In the gel images ( Figure 2A ), the shift was observed only in the presence of specific nucleic acid effector E1 but not in the absence of E1 for both hairpin oligonucleotides HP1 and HP2. This indicates that the binding of thrombin to hairpin oligonucleotides is weak when hairpin oligonucleotides assume the stem-loop structure, thus allowing the antidote to bind to the aptamer. However, the binding with thrombin is significantly enhanced when hairpin oligonucleotides hybridize to effectors, thus freeing the aptamer from the antidote. The intensity of different bands in the gel images was used to determine the fraction of hairpin oligonucleotides HP1 and HP2 bound to thrombin, which was plotted against the concentration of effector E1 in Figure 2B . With the increase of E1 concentration, the fraction bound to thrombin increases for both HP1 and HP2. This demonstrates that the binding between hairpin oligonucleotides and thrombin can be regulated by the specific nucleic acid effector in a concentration-dependent manner. At the same concentration of E1, more HP2 binds to thrombin than HP1 does because the spacer between the sensing loop and the aptamer in HP2 makes it easier for the aptamer to fold into the correct configuration. For an aptamer to bind with its target in a high affinity and specificity, it is crucial for the aptamer to assume the correct configuation.
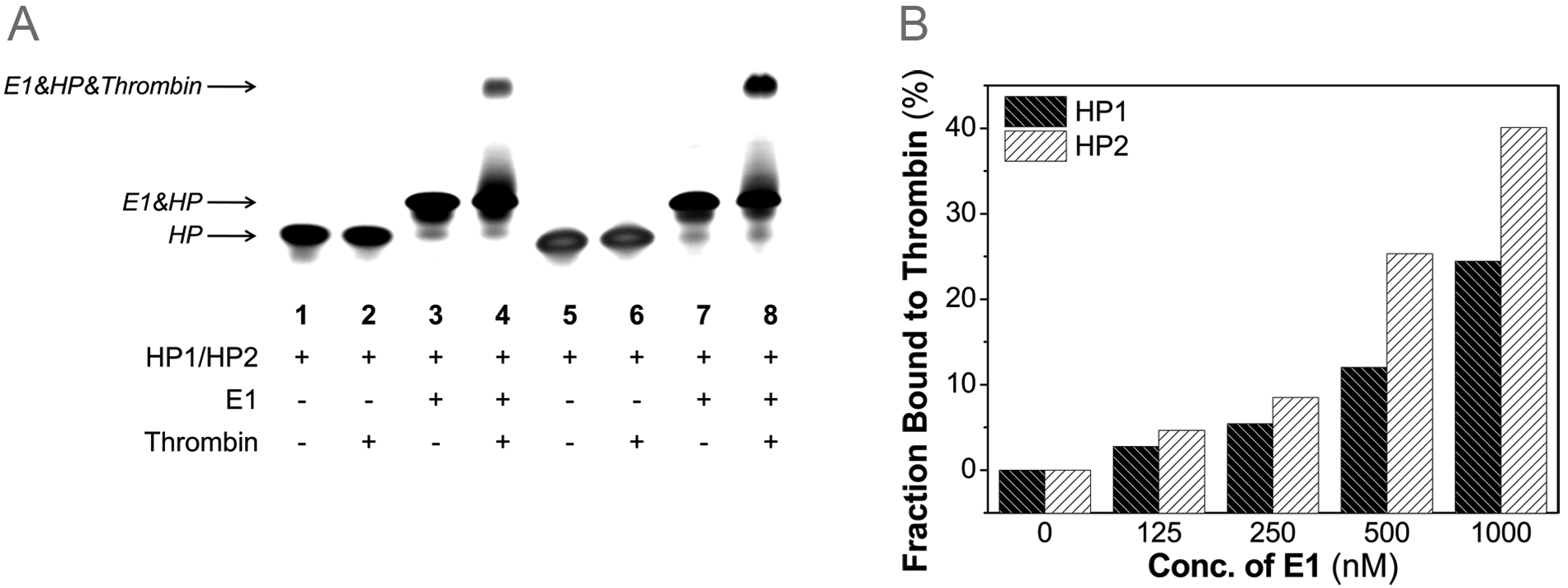
Binding of HP1 and HP2 to thrombin. (
Activity Tests
Two classes of substrates are commonly used for measuring the activity of thrombin: synthetic peptide substrates and natural protein substrates (e.g., fibrinogen). The synthetic substrates typically consist of a peptide that can be specifically cleaved by thrombin and a chromogenic or fluorogenic group that will be released upon cleavage of the peptide. These synthetic substrates, especially the fluorogenic peptide substrates, 28 have enabled the real-time monitoring of thrombin activity in a wide range of samples. However, these small peptide substrates, which interact with the active site of thrombin, 29 are not suitable for characterizing the inhibition of thrombin activity by the 15-nt DNA aptamer used in the present study. This is because this aptamer inhibits the activity of thrombin by competing with fibrinogen for the anion-binding exosite I on thrombin.30,31 Therefore, a fibrinogen clotting assay, which is based on the increase in the solution absorbance due to thrombin cleavage of fibrinogen, was used here. As shown in Figure 3A , the change rate of absorbance at 400 nm increases with the thrombin concentration, and the linear regression results in a R2 of 0.979. This indicates that the change rate of absorbance is well correlated with the thrombin activity. Figure 3B further demonstrates that the fibrinogen clotting assay based on the absorbance change can be used to quantify the inhibition on the thrombin activity by the original 15-nt thrombin-binding aptamer (TBA). Moreover, compared with the conventional assay that is based on the clotting time from a fibrometer, the assay here could potentially be used to monitor the thrombin activity in real time because the increase in the solution absorbance can be continuously measured.
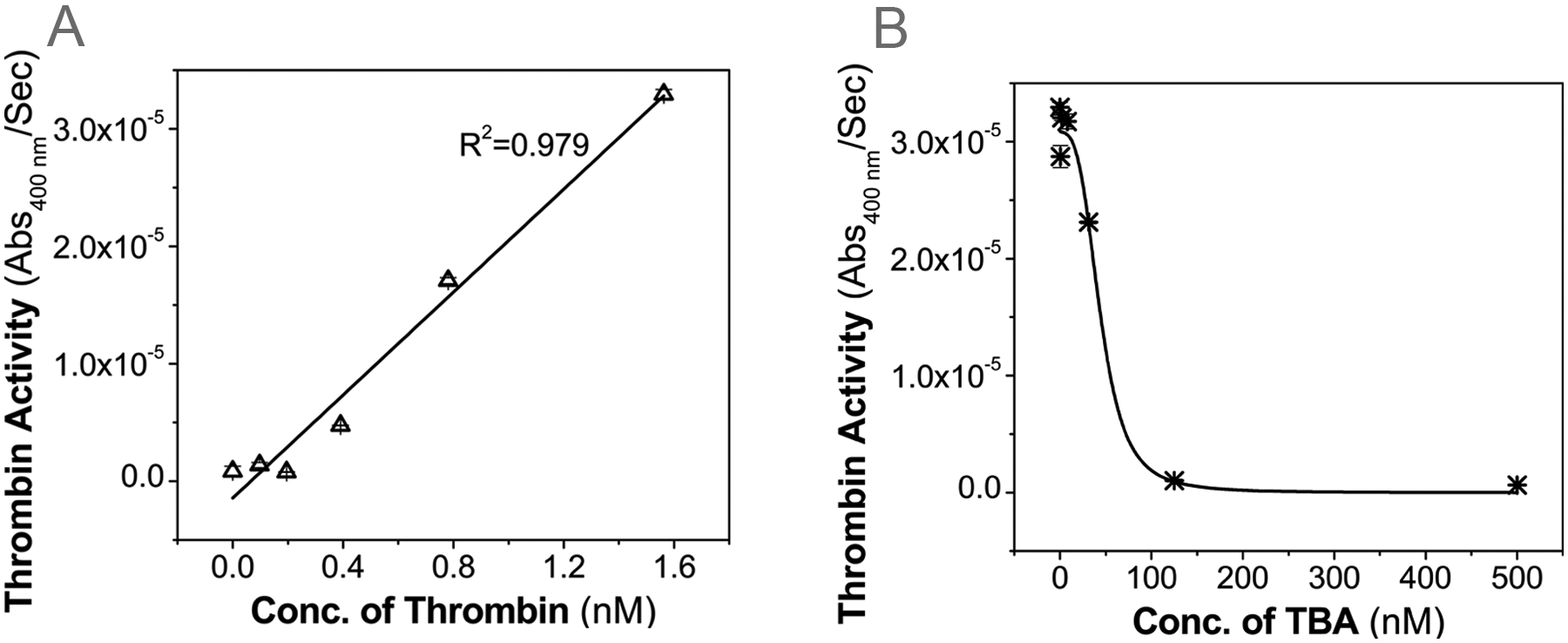
Fibrinogen clotting assay based on the change in the solution absorbance. (
The nucleic acid effector E1 has been shown to increase the affinity between hairpin oligonucleotides (HP1 and HP2) and thrombin in the gel shift assay. Therefore, it is expected that E1 will increase the inhibition of HP1 and HP2 on the thrombin activity. This was confirmed with the fibrinogen clotting assay in Figure 4A . The activity of thrombin exhibits a decreasing trend with the increase of E1 concentration when there is HP1 or HP2. In the control experiment with 1× binding buffer, the decrease is nominal. This suggests that the effector itself does not interact with thrombin and instead facilitates the interaction between hairpin oligonucleotides and thrombin by binding to hairpin oligonucleotides. Comparison of HP1 and HP2 in their thrombin-inhibiting ability further reveals the importance of an appropriate spacer between the sensing loop and the aptamer stem. Figure 4B implies that there is great room for improvement on the design of spacers. Alternative spacers, such as polyethylene glycol (PEG), which has been used to maintain the activity of aptamers in various applications requiring labeling or sequence alteration of aptamers,14,32 are expected to significantly reduce the concentration of the effector-responsive hairpin oligonucleotides required to achieve a certain level of therapeutic activity.
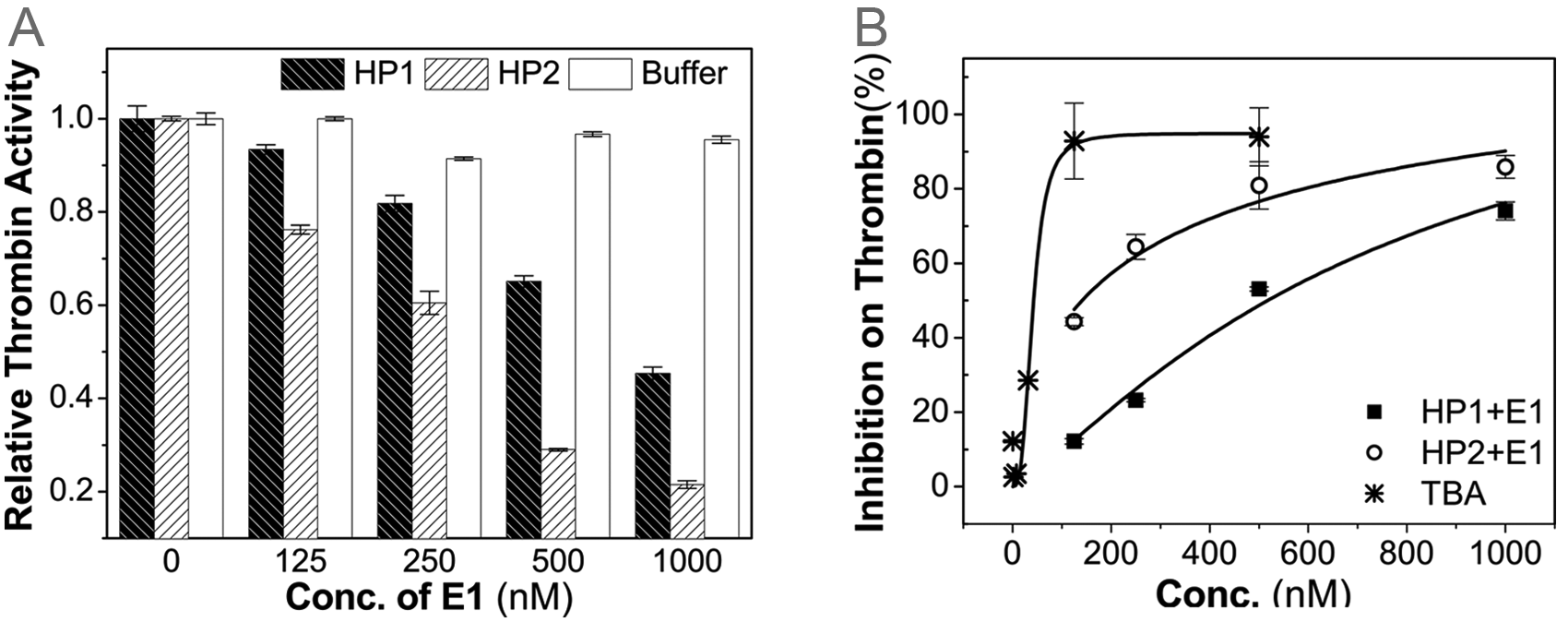
Inhibition on the fibrinogen-clotting activity of thrombin. (
Specificity and Reversibility
The above results from the binding and activity tests have proved that HP1 and HP2 can be regulated by the effector E1, but it is still unclear whether the regulation is specific and reversible. The specificity was examined by the introduction of mismatched effector sequence E1-C ( Figure 5A ). Compared with the perfectly matched effector, the inhibition on thrombin was only nominal. This implies that the inhibition by HP1 and HP2 relies on the recognition of a specific effector by the sensing loop. The reversibility was demonstrated with E2 and E2-C in Figure 5B . The 30-nt E2 contains a 20-nt region that is complementary to the sensing loop of HP1 and HP2, as well as an additional 10-nt region that is irrelevant to HP1 and HP2. E2-C is a 30-nt oligonucleotide that is completely complementary to E2. Therefore, E2-C has a higher affinity to E2 than HP1 or HP2 does. In the experiments, equal molar ratios of E2 and HP2 were first mixed with thrombin and fibrinogen. Then, E2-C was added to remove E2 from HP2. Last, excess of E2 was introduced to provide additional E2 accessible by HP2. It can be seen that HP2 was initially in the “activated” state when an equal molar ratio of effector E2 was present, then deactivated when most of E2 was occupied by E2-C, and sequentially activated when additional E2 became available. This suggests that the effector-responsive hairpin oligonucleotides can reversibly adjust the aptamer activity according to the changes in the concentration of nucleic acid effectors.
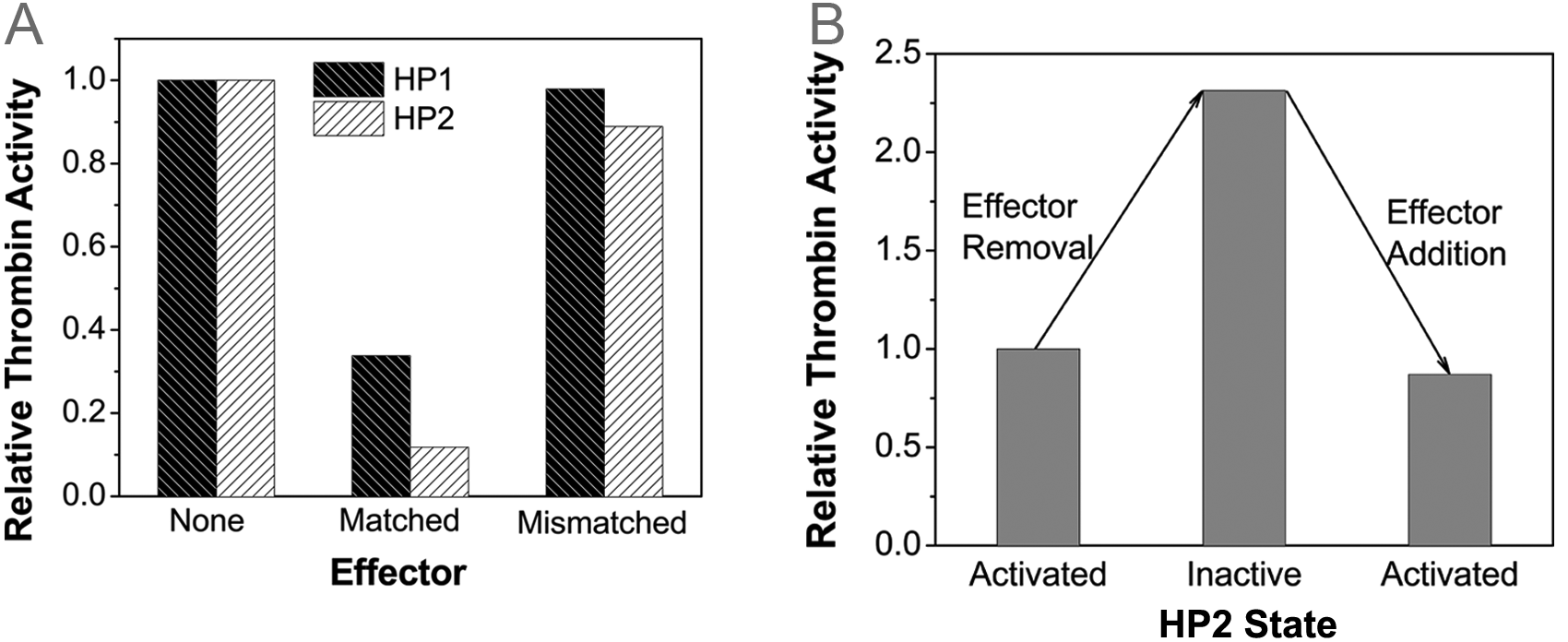
Specificity and reversibility. (
Discussion
In conclusion, we developed an alternative strategy to regulate the aptamer activity according to the specific nucleic acid effector. Exploiting the spontaneous conformational change of hairpin oligonucleotide accompanying the specific molecular recognition eliminates the need for additional means to convert the recognition event into the change of aptamer activity, such as the irreversible cleavage capability of nucleic acid enzymes used by some existing strategies. Therefore, in addition to the specificity, which is intrinsic to the strategies where the regulation is triggered by the specific molecular recognition, this strategy also has the unique advantage of reversibility.
Although the concept of the strategy was demonstrated here with a particular aptamer (the 15-nt antithrombin aptamer) and oligonucleotide effectors in the test tube, it should be of general applicability to other aptamers and nucleic acid effectors (e.g., mRNA and DNA). Our ongoing efforts to optimize the design of effector-responsive hairpin oligonucleotides include minimizing the remaining activity in the “inactive” state and fully restoring the activity of the original aptamer in the “activated” state. With ongoing optimization and future intracellular tests, this strategy could potentially be used to create “on-demand” aptamer therapy. The potency of the therapy (the aptamer activity) is continuously adjusted based on the disease status that is indicated by the amount of effectors (mRNA or DNA biomarkers).
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported was supported by University of Miami through Provost’s Research Awards and National Institutes of Health through the NIH Roadmap for Medical Research (PN2 EY018228).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.